|
|
酵素純化 Enzyme Purification 2 色層分析法 Chromatography |
總目錄 ■ 上課網 ■ 下載區 ■ |
|
|
參考資源 |
|||
|
目 錄 |
2.1 色層分析原理 2.2 膠体過濾法 2.3 離子交換法 2.4 親和層析法 2.5 HPLC 及 FPLC |
典型的葉綠素色層分析圖
(Wikipedia CC by Florian Siebeck) |
下載 [內文 pdf] [投影片 pdf] {BCbasic} 連結生物化學基礎
|


|
2 色層分析法 |
參考資源 |
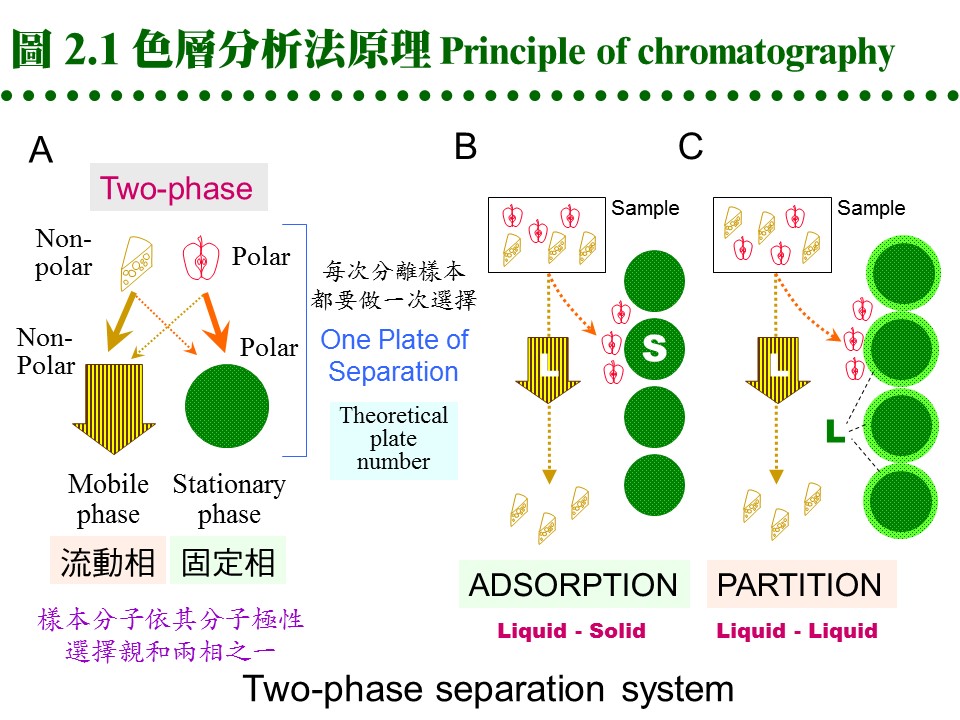
2.1 色層分析原理:a. 系統組成: 每種層析系統均由兩個「相 phases」組成,主要分成 固定相 (stationary phase) 及 流動相 (mobile phase),二者各有不同的極性或非極性強度。而樣本分子因自身極性的強弱,與此二相之親和力不同:與固定相之親和力較大者,易留滯原地;與流動相之親和力大者,則易隨流動相移動,因而達到分離的效果。圖 2.1 以圖解說明此一機制。
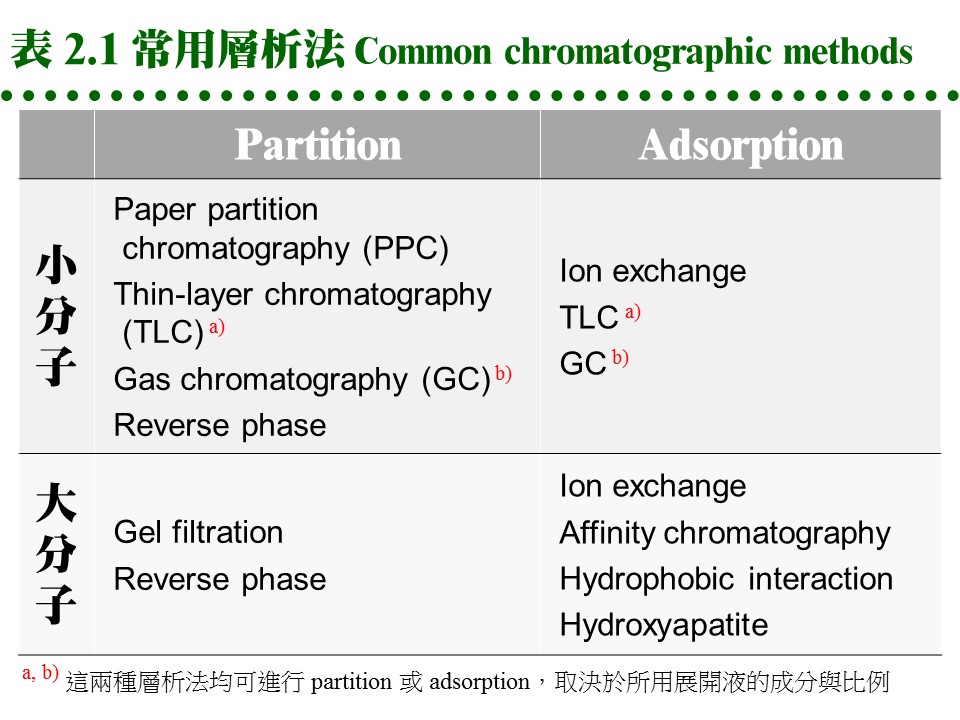
(每張圖表均連結有 960 x 720 清晰版本) 這種親和力的產生,決定於樣本或兩相物質之化學本質,是屬極性或非極性,進而遵循『Like dissolves like』的原則;即極性分子較易溶入極性的固定相或流動相,非極性分子則易溶入非極性者;因此一個樣本分子,就依其極性大小在此兩相間做選擇與分離。 c. 方式很多: 因為固定相或流動相可能是 固 (S)、液 (L) 或 氣 (G) 相之一,故可有多種組合方式: Partition chromatography: 固定相 (L) 流動相 (L) 例:PPC, TLC, 膠体過濾 Adsorption chromatography: 固定相 (S) 流動相 (L) 例:TLC, 離子交換 Gas-liquid chromatography: 固定相 (L) 流動相 (G) 例:GC d. 常用層析方法:
|
[History] |
| ▲ | |
|
[PPC] [TLC] |
|
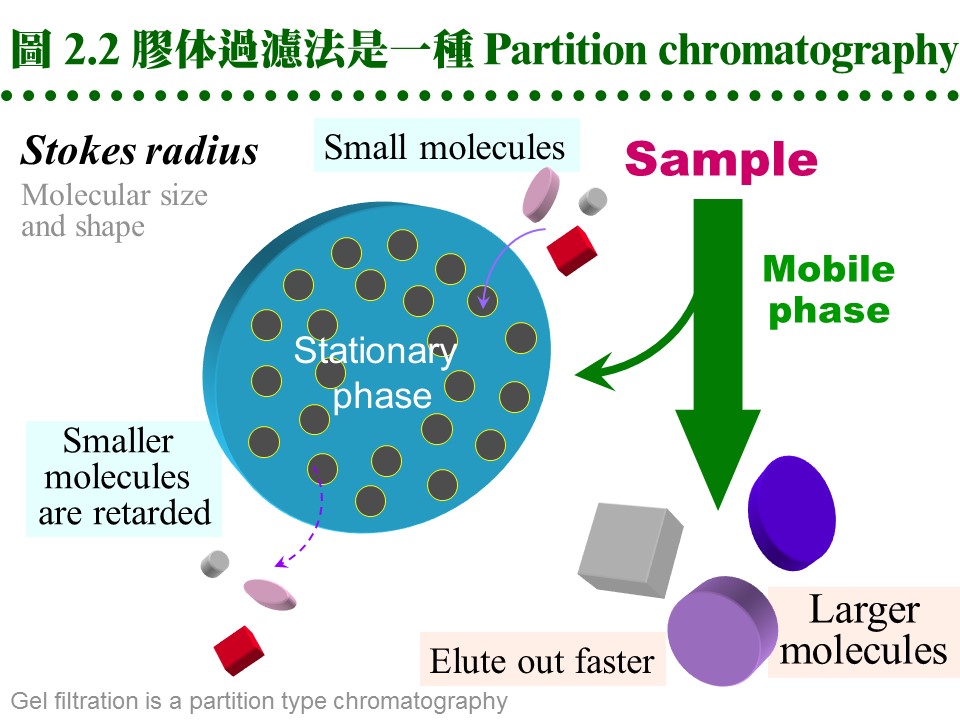
2.2 膠体過濾法:2.2.1 原理概述: 膠体過濾屬 partition 層析法,流動相為溶離緩衝液,固定相為膠体孔隙內的緩衝液。溶質 (樣本蛋白質) 根據其分子量的大小,決定分佈在這兩相的比例。分子量大的不易進入膠球,隨流動相溶離;分子量小的,則易竄入膠球內的固定相,而被延滯流出膠柱 (圖 2.2)。分子的 形狀、大小 均為影響因素,即與其 分子半徑 (Stokes radius) 有關,與分子量不完全成正比關係,但一般視蛋白質為球形,其形狀影響暫予忽略。
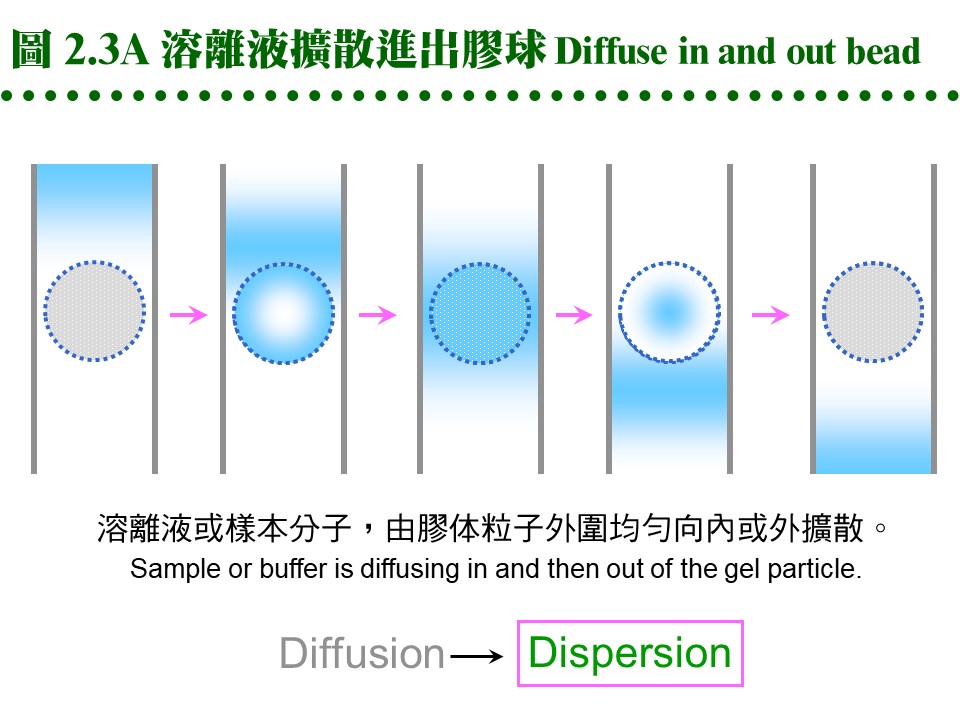
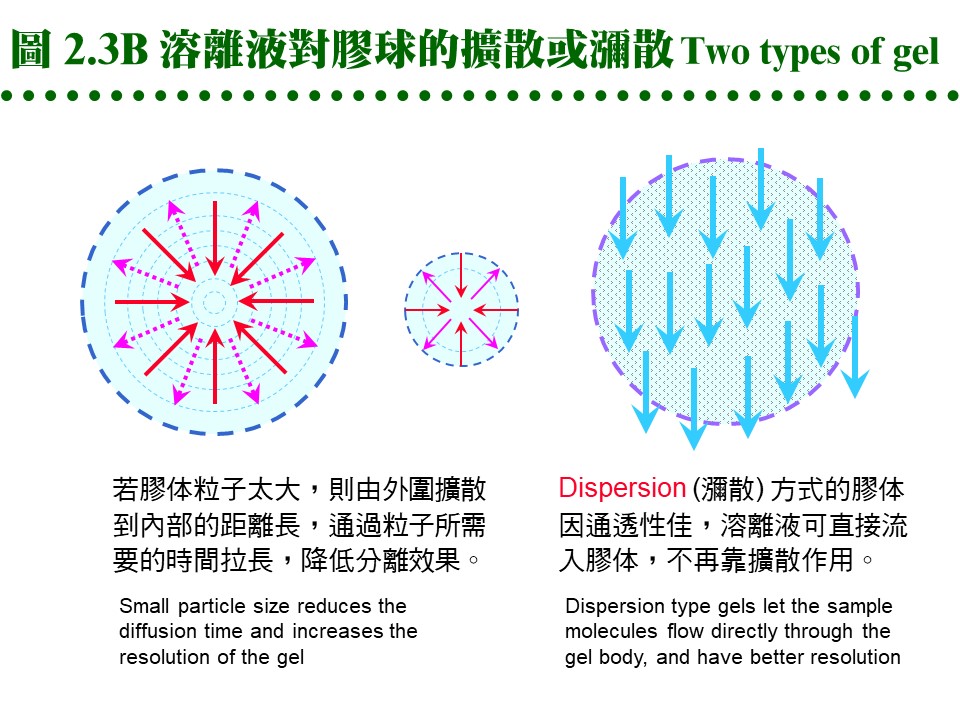
2.2.2 膠体介質 (support, matrix): 膠体介質是三次元的網狀小球,由長鏈聚合物交織而成,有三大類不同材質。 a. Dextran: 是葡萄糖組成的多醣長鏈,經過架橋修飾,成為內部有均勻孔道的小球。早期由瑞典 Pharmacia 開發的最基本產品形式有下面數類: (1) Sephadex G 系列:是最先推出的介質,有各種適用分子量範圍,如 G-10, 15, 25, 50, 75, 100, 150, 200 等,數字越大,表示膠球孔徑越大,其適用的分子量就越大。要注意 G-150 以上的膠体,在高壓下會被壓垮而使流率變慢,甚至無法流通,儘量改用 Sephacryl 或 Sepharose 系列。G-25 以下者,常做為蛋白質 脫鹽(desalting) 之用。 (2) Sephacryl S 系列:額外使用 Bis 做架橋支持,因此比 Sephadex 有更好的耐高壓能力。有S-200, 300, 400, 500, 1000等不同孔徑,適用分子量大的樣本,但注意它有很高的非專一性吸附,要小心蛋白質被『吃掉』! 最好用鹽濃度較高的溶離緩衝液 (0.2 M NaCl)。 (3) Sephadex LH 系列:Sephadex 上的 OH-基團被修飾成 hydroxylpropyl 衍生物,成為 LH 系列,疏水性較大,可兼用在極性或非極性緩衝液。 b. Agarose: 洋菜醣是由海藻抽出的直鏈聚醣,長鏈分子間以氫鍵架橋,形成三次元膠体,可以濃度來控制孔徑大小。故有些 agarose 材質的膠球,不能加高熱,否則會融成一塊膠片 (如培養用的洋菜膠);用在分子量特別大的分子 (如核酸) 或粒子 (如病毒)。 (1) Sepharose 及 Sepharose CL 系列:Cross Linking 系列特經架橋反應補強,可耐高壓高溫。各有 2B, 4B, 6B 三種,數字表示含膠百分比,數字越大孔徑越小,與 Sephadex 相反。 (2) Bio-Gel A 系列:Bio-Rad 有 A-0.5M, A-1.5M, A-5M, A-15M, A-50M, A-150M,數字表示其所適用的最高分子量,以百萬為單位。 c. Polyacrylamide: 像電泳膠体一樣,由 acrylamide 聚合成固定大小孔徑,製成小球,供層析分離之用。商品為 Bio-Gel P 系列 (Bio-Rad),有 P-2, P-4, P-6, P-10, P-30, P-60, P-100, P-150, P-200, P-300等,最高可使用在分子量 300,000 者,高壓之下的流速亦會變慢。 d. 膠球大小: 膠球粒看起來都很細,但都有一定的大小,一般可分為 coarse, medium, fine, super fine 四種粗細 (grade);越粗的膠体,流率越好,但解析力越差。因膠球外面的緩衝液是由膠球表面向內擴散,樣本蛋白質也是以相同的擴散方式進入膠球,再由中心向外擴散出來,因此膠球半徑越大,擴散距離越大,效果就越差。圖 2.3 A 說明這種傳統擴散式介質的機制。
而近年來因材料科學的進步,發展出通透性特強的膠球,緩衝液可直接浸潤而進入膠球,不需經擴散作用,稱為 dispersion 瀰散式的膠体 (圖 2.3B),效果較好且快速。
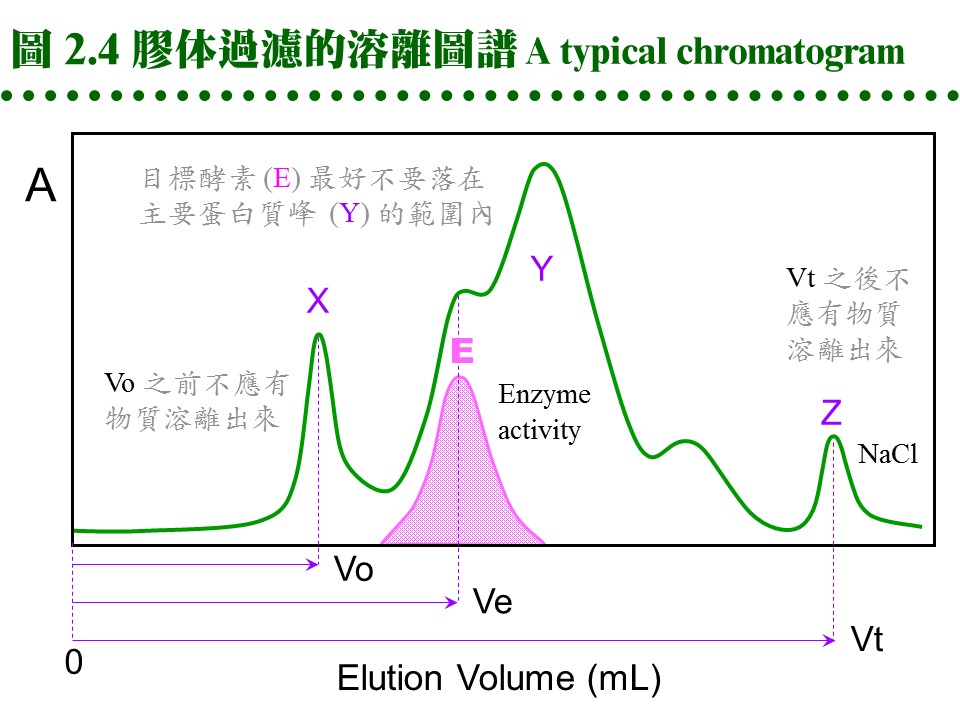
2.2.3 膠体管柱: a. 管柱性質: 膠体過濾都以管柱方式進行,不論使用何種介質,管柱的性質大致可以如下預測 (圖 2.4):當管柱裝填完成,若膠柱總体積為 100 mL (total volume, Vt),則膠柱內的液相總体積約 90 mL (liquid volume, Vl),其中流動相的体積 (即介於膠球之間的緩衝液總体積, void volume, Vo) 約為 35 mL,樣本分子則應於 35~90 mL (Ve) 間溶離出來。如同 TLC 的 Rf 值,膠体過濾也有表示樣本溶離程度的指標 (Kav);樣本的 Kav 與分子量成反比,因此可用來測定分子量,現在多直接以溶離体積表示樣本溶離程度。
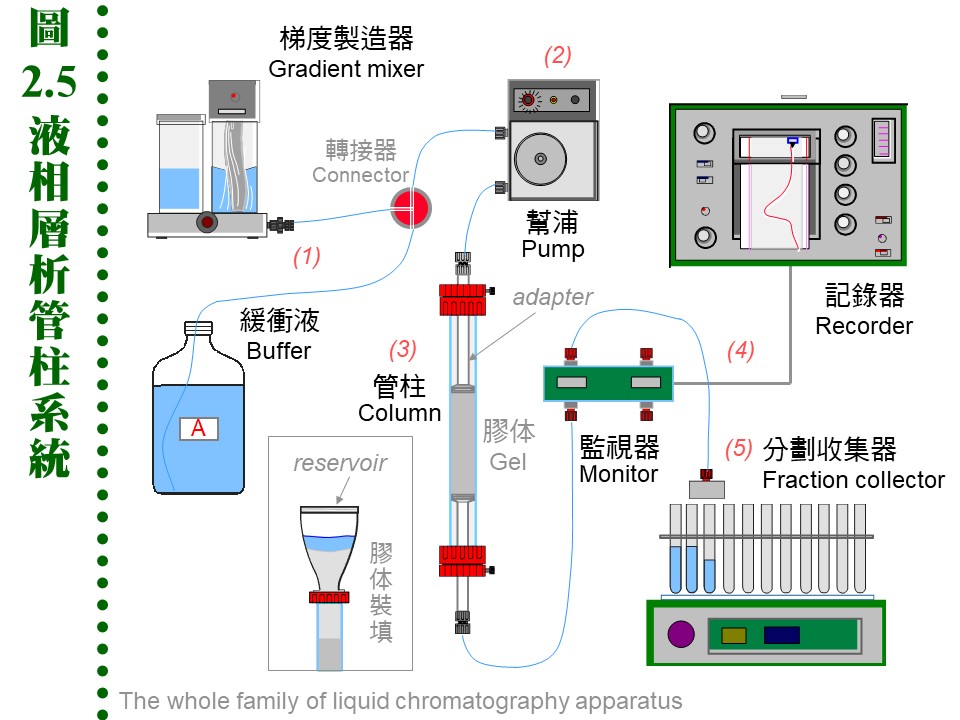
b. 內在因素: 膠体過濾法在操作時,有些內在的問題,會影響結果的好壞: (1) 擴散及亂流:由於兩相都是在液相中進行,樣本在膠柱中的 擴散現象 相當顯著;又因液体在膠球間流動時,受 重力 及 對流 的影響,會造成 亂流。擴散及亂流都會使膠体過濾的解析力大幅降低。 (2) 管柱設計不良:經常是致命的傷害,例如 無效空間 (dead volume, 見 2.3.4b) 過大、緩衝液進入膠体時流動不均、溶離液出口端的管路太長或太粗等。 c. 解決擴散及亂流: 若降低 膠球粒子 的大小,則可改善之;因此越細的膠球,其解析力就越佳。但膠体若太細,會造成流速下降,則要用更大的壓力進行溶離,許多膠球耐不住如此高壓。因此若補強膠体材質,以架橋來支持膠体構造,或改用矽膠質為材料,可改善流率並增加解析力,即為 HPLC 系統 (2.5)。使用上述 dispersion 式膠体,也是可行方法。 d. 膠体的選擇: 取決於所要分離蛋白質樣本的分子量大小,並且預期溶離出來的蛋白質峰,可出現在 Vo-Vt 區間的前半段,以降低因擴散所造成的不利影響 (使目標蛋白質早些溶離出管柱)。通常分子量大於十萬可用 Sepharose CL 系列,小於五萬者用 Sephadex G-100 以下,其間則使用 Sephacryl S-200 或 300。避免使用 Sephadex G-150 或 200,因其流率不佳;其它廠牌相對應的產品,亦可使用之。 e. 管柱大小: 視所要分離樣本的体積而定,一支膠球体積為 100 mL 的管柱,可分離 1~3 mL 樣本。膠体過濾管柱以細長較妥,通常直徑為 1.6 或 2.6 cm,長度 80~100 cm,太長者擴散作用明顯。離子交換法及大量生產時的製備式管柱多使用矮胖型,以增加流率。 f. 管柱系統: 完善者包括下列各部分 (圖 2.5): 緩衝液 (1) → 幫浦 (2) → 管柱 (3) → 監視器 (4) → 分劃收集器 (5) 其中監視器並非必需,管柱種類很多,上等備有 adaptor 可降低 無效空間 (2.3.4b),且使用很方便。通常要在冷房中操作,因此儀器的維護要更小心。取出冷房後要立刻在乾燥環境下烘乾回溫,以免潮濕造成短路或發霉。梯度製造器使用在離子交換法。
2.2.4 管柱操作: a. 膠体處理: 許多膠体是乾燥粉末,使用前要先行膨潤,如 Sephadex, Bio-Gel P;其餘如 Sephacryl, Sepharose (CL), Bio-Gel A均為已膨潤者,直接在玻璃漏斗以緩衝液洗過 (圖 2.6A) 即可裝填;若有需要,可以減壓幫浦抽去氣泡。注意 Sepharose 及 Bio-Gel A 不可加熱或高壓滅菌。由管柱大小算出所需膠体的体積,再加一成,準備裝填管柱。
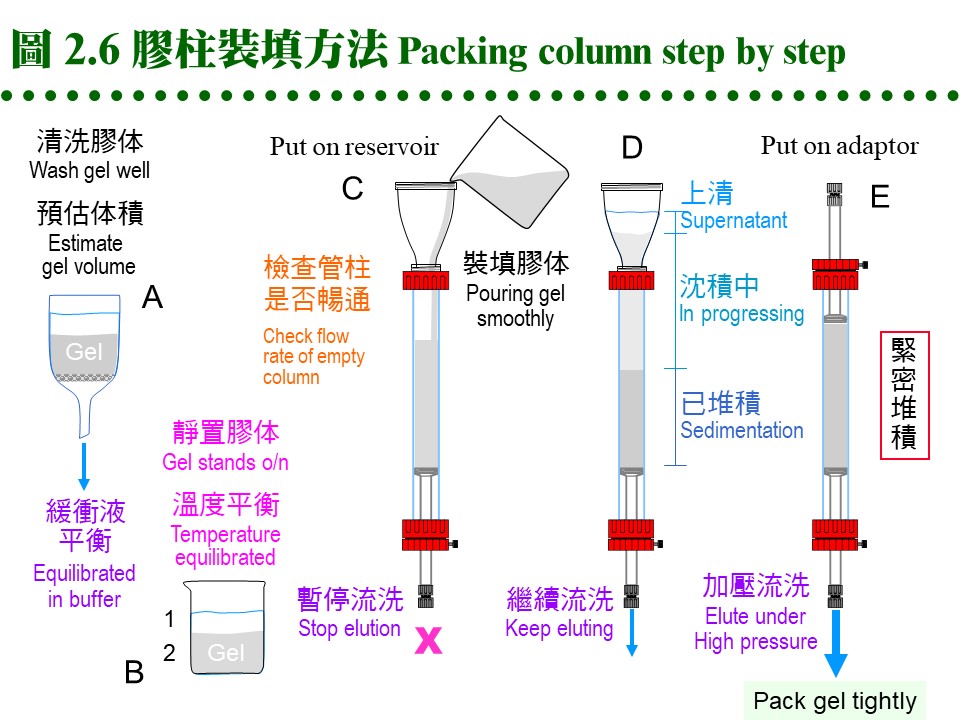
b. 膠体裝填: 以下是裝填膠柱的詳細步驟,最好實地觀看示範操作或教學影帶。 (1) 洗好的膠体浸在緩衝液中,靜置過夜使之沉降,把上清部份的体積大致調為膠体体積的一半 (圖 2.6B 膠体佔三分之二)。 (2) 先把各儀器連結好,確定可正常運作,架好管柱,注意要確實垂直地面。 (3) 若要裝填較高的膠柱,則要裝上 reservoir,否則一次倒滿只能沉降到管柱的七成高。 (4) 先在管柱內加一些緩衝液 (約 5 cm 高),看能否順利流出,再關住出口。 (5) 把上述膠体攪拌均勻,成為懸濁液,但勿產生氣泡。 (6) 沿著管柱的管壁,慢慢倒入膠体;勿粗魯灌入,避免生成氣泡 (圖 2.6C)。 (7) 一口氣倒完後,等約 1 min 後打開出口,膠体開始沉降。 (8) 不久整隻管柱分成三層 (圖 2.6D),最上為澄清的緩衝液,下層為已堆積好的膠体,顏色較白,中層為沉降中的膠体。勿使上方緩衝液乾掉。 (9) 待膠体完全沉降後 (中間層消失),應得到預計的膠体高度,否則要追加或挖去膠体;要添加膠体時,先把膠柱上方約 5 cm 高的膠体均勻懸濁後再加入。 (10) 除去 reservoir,加上 adaptor,注意系統中不能有氣泡 (圖 2.6E)。這個步驟較易出問題,請仔細研究清楚所有細節,小心練習好才進行。 (11) 連通整個系統,檢查系統的封閉性,調整幫浦流速。 (12) 以較高的流速洗 (45 mL/h, 約 150% 流速),以平衡完全。通常膠体裝填時的流率,要比操作時稍高;Sephacryl 則要更高流速,但當使用 Sephadex G-100 以上者,只能用平常流速,否則膠体會被壓垮。 (13) 膠体高度可能會稍微下降,則 adaptor 要再往下壓,以完全接觸膠面。 (14) 以正常流速 (約 30 mL/hr) 流洗數小時,可洗過夜,同時準備樣本。 c. 樣本体積: (1) 樣本体積限定在膠柱体積的 1~3%,可加入甘油以增加密度。 (2) 裝有 adaptor 的管柱較方便,可用幫浦注入,否則要直接把樣本加在膠体表面上,先吸去膠柱上面的緩衝液後,再小心注入樣本 (乾式);或不吸去緩衝液,把密度較大的樣本,在緩衝液中直接加入,讓它自動沉降在膠体表面 (溼式)。 (3) 樣本溶液不可有沉澱,否則要先離心除去之,太濃或太稀均不適宜。注入樣本應極為小心,勿破壞膠体表面! d. 溶離速度: 以直徑 1.6 或 2.6 cm 管柱而言,通常每小時流速約 30 mL 左右,較粗的管柱可加快,Sephadex G-150 或 G-200要減慢,而 Sephacryl 溶離速度可加快一倍。流出液約每 2-5 mL 收集一個分劃,但可依情況自行增減,最好使用分劃收集器,讀滴數、秒速均可。要注意勿讓膠体乾掉,也要小心收集器很容易出毛病,沒有收到樣本而流失。 e. 管柱保存: (1) 膠体管柱暫不使用時,可在緩衝液中加 NaN3 (0.01%) 流洗一次,關好出口。 (2) 長期不用時最好取出膠体,在玻璃漏斗中以 PBS 洗過數個体積後,保存在 4℃中,再加數滴 NaN3 防菌。 (3) 若發覺膠体太髒,可用 0.2 M NaOH 或 NaCl 先洗過,再以緩衝液平衡;再度取出使用時,要注意有沒有長霉 (黑色棉絮狀小球),膠体有無結塊。 (4) 已膨潤的膠体應貯於 4℃,切勿貯藏在零下的溫度,膠体的結構會被冰晶破壞;乾粉或未尚未開封者,可貯於室溫。 (5) 膠体外表看來都一樣,一定要標示好,以免混淆不清;千萬不要把兩種膠体混在一起,或者弄錯標籤,結果都會很淒慘! 2.2.5 問題及解決: a. 管柱裝填: 膠柱是否良好,可跑 Blue Dextran (Pharmacia) 試之,藍色色帶應平穩地往下移動,色帶厚度會稍加寬,但不該有拖尾、變斜,甚或成為不規則亂流!也可用手電筒在管柱後方打光,看膠体中有無氣泡。 b. 溶離緩衝液: 流速太快會造成分離結果不好,通常是色帶拉長或呈現不規則。緩衝液中的離子濃度有相當影響,通常不能用蒸餾水來溶離。樣本分子在通入膠体後不久,其緩衝液即被管柱中的緩衝液所取代;若此二種緩衝液不同,則因為離子濃度的改變,某些蛋白質可能會 鹽析 出來,沉澱在膠面。 c. 活性消失: 有些樣本蛋白質,需要金屬離子、輔酶、輔因子等小分子,共同達成其活性,在通過管柱後,可能被排除而失去活性。可在活性分析時補充,或在溶離緩衝液中添加。若蛋白質的回收量太低,要注意膠体有無吸附現象。 d. 使用溫度: 膠体管柱由冷房移到室溫後,會漸生成小氣泡,不能再用。反之由高溫處移至低溫處時,則無此問題。緩衝液也有同樣現象,應當注意。 e. 老舊膠柱: 管柱經長期未使用,要注意有無長霉,管柱有無乾裂 (用手電筒檢查)。使用太多次數後,膠柱最上方的表面會有沉澱或變得較髒,可稍挖去表層,再小心輕輕攪散,再讓膠体表面重新沉降平整,對結果影響不大。
|
|
| ▲ | |
|
|
|
| ▲ | |
2.3 離子交換法:離子交換法乃是利用分子帶電性質的差異來進行分離,解析力好且具多樣性,是常見且應用極廣的純化方法。 2.3.1 原理概述: a. 離子交換法: 是一種 adsorption 層析法,流動相為溶離緩衝液,固定相為介質擔体表面的帶電基團。樣本中的各種離子,與介質表面帶電基團間的親和力強弱不同,吸附上去之後,可使用不同離子濃度的緩衝液,分別溶離出這些成分 (圖 2.7)。
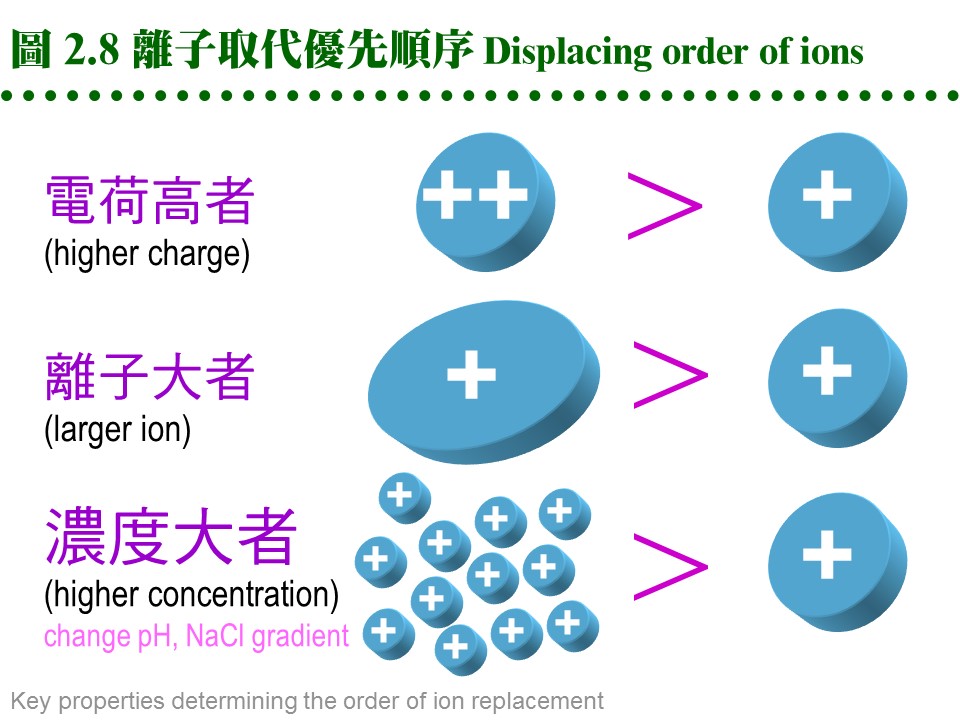
b. 兩大類離子交換介質: 由介質帶電基團的不同,可分為兩大類: 介質-帶電基團 (counter ion) (1) 陽離子交換介質 (cation exchanger): 擔体-陰離子基團 ..... 陽離子 (2) 陰離子交換介質 (anion exchanger): 擔体-陽離子基團 ..... 陰離子 c. 離子取代順序 pecking order: 離子交換的進行,可視為各種 counter ions 間,對擔体介質上帶電基團的爭奪戰,離子 (包括蛋白質) 競爭結合到固体介質上;其競爭優勢順序如下 (圖 2.8): (1) 帶電荷高者取代帶電荷低者。 (2) 電荷相同時,原子序 (或離子体積) 大者優勢。 (3) 濃度 可克服以上兩種優勢,高濃度之氫離子可取代其它陽離子。
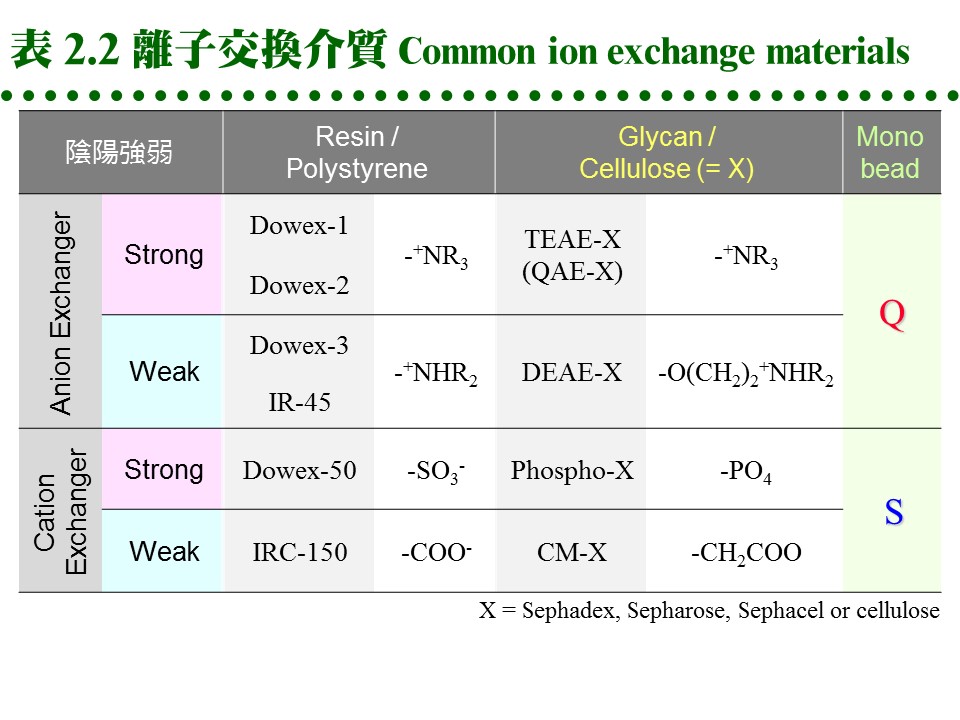
d. 離子取代優先順序: 陽離子:兩價陽離子 > NH4+ > K+ > Na+ > H+ > Li+ 陰離子:I- > Br- > Cl- > HCO3- > CH3COO-, OH- 2.3.2 離子交換介質: a. 介質種類: (1) 離子交換介質的種類很多,歸納起來分為 陰離子 及 陽離子 兩大類;每一類又依其帶電基團的強弱,分為 強、中、弱 三種。 (2) 另外依介質的材質不同,略分為 合成樹脂 (resin) 及 聚醣 (glycan) 兩種,前者對蛋白質的純化並不適用,只可用在小分子樣本。 (3) 聚醣多使用 Sephadex, Sepharose, cellulose 等為擔体,在糖分子加上帶電基團;而 cellulose 又有做成結晶球形的 Sephacel,可增加膠柱的流率。 (4) 因為材料科學的進步,產出了許多新的材質,更為堅硬可耐高壓,且所能吸附的樣本容量也大增。Pharmacia 推出新一代介質 Monobead,也分有陽離子 (Q) 及陰離子 (S) 兩種,都是預先裝填在管柱,直接接上 FPLC 系統使用。
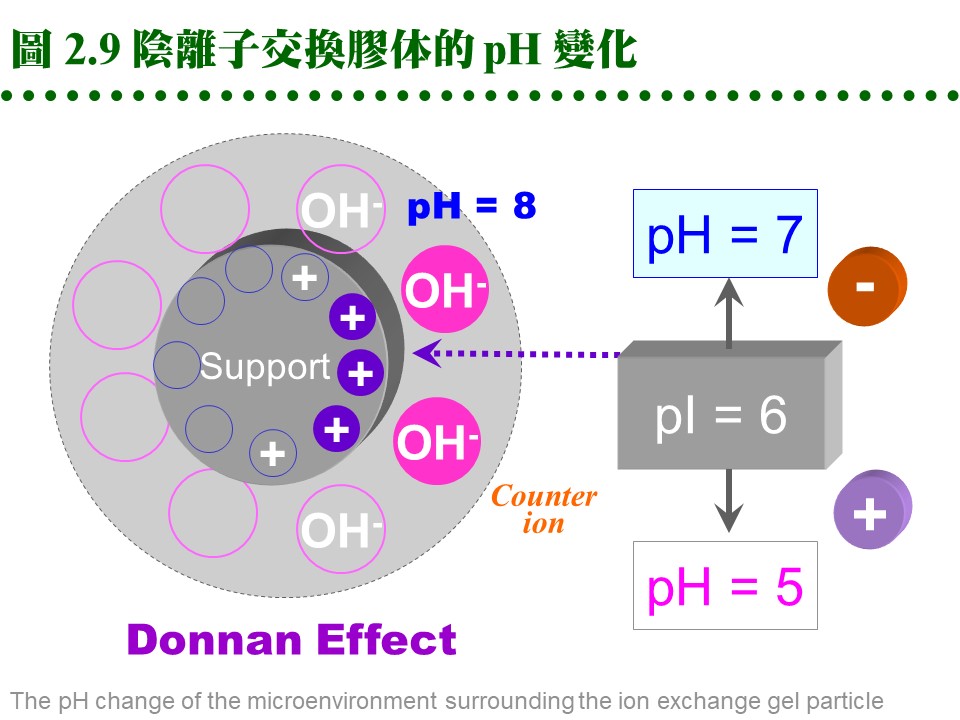
b. 選擇交換介質: (1) 若已知樣本蛋白質的 pI,則先選擇所要使用緩衝液的 pH,使蛋白質帶有正 (或負) 電,而採用陽 (或陰) 離子交換介質。 (2) 若不知樣本的 pI,則可取一系列試管各裝少量介質,在各種緩衝液 pH 下,加入適量的樣本蛋白質,然後測上清中有無酵素活性殘留,得知樣本蛋白質有無吸附上去,就可選擇適當的介質及緩衝液 pH。 c. 一般使用: (1) 通常在純化蛋白質時,都使用較弱的離子交換介質,如 DEAE (diethylethanolamine) 或 CM (carboxymethyl);介質則用聚醣類為材料,多使用Sepharose CL-6B 或 Sephacel,而不用体積變化很劇烈的 Sephadex,或流率較差的 cellulose。 (2) Sepharose 本來有膠体過濾的作用,應用在離子交換時,作用並不明顯;但在分離異構酶時,因各個異構酶的分子量極為相近,避免使用之。 (3) DEAE 型膠体使用的 pH 不能高過 9,CM 者不能低於 pH 6,否則介質會失去原先帶有的電荷。 d. 介質容量有限: (1) 離子交換介質與蛋白質的結合總量有一定的限度,稱為該交換介質的 容量 (capacity);若超過此一容量,多出的樣本會直接流出。 (2) 交換介質的結合容量大小,受層析條件不同、蛋白質種類不同、緩衝液不同、pH 或離子濃度不同等,有很大的差異。如 DEAE-Sepharose CL-6B 每 100 mL 可結合 11 克白蛋白,但對 ferritin 只有 0.4 克。 e. 介質表面的微環境: (1) 由於交換介質的帶電性,其微視環境中的 pH,並不呈均勻的狀態。 (2) 緊靠介質表面的 pH,要比外圍緩衝液 pH 相差一個 pH 單位左右:陰離子交換介質高一個單位,陽離子低一個單位 (稱為 Donnan effect),可參見圖 2.9。
f. Hydroxylapatite: (1) 可與 DNA 或 RNA 結合,原本用在分離單股與雙股 DNA,是一種結晶型的磷酸鈣,其作用機制不很清楚,但顯然與其帶電性質有關。 (2) 操作法與離子交換法類似,在低離子濃度時使蛋白質結合上去,再以高濃度溶離下,但較複雜;不同的鹽類 (如 磷酸鹽, NaCl 或 CaCl2),會有不同的溶離結果,要以實驗嘗試求得。 2.3.3 緩衝液與層析系統: a. 緩衝液種類: 可能會影響離子交換結果,例如 DEAE 介質若使用磷酸緩衝液,則其中的磷酸離子 (帶兩個負電) 與交換介質的結合力相當強,會影響樣本蛋白質的結合。但反過來說,此時能夠結合上去的樣本蛋白質,就有相當的強度。 b. 緩衝液 pH: (1) 緩衝液的 pH 可定在樣本蛋白質 pI 的上或下一個 pH 單位 (參見圖 2.9),使樣本分子帶有正確的電荷,能夠結合到所選用的介質上去,但又不會太強,以免難以溶離下來。 (2) 用酸鹼度溶離時,當緩衝液的 pH 趨近樣本分子的 pI 在 0.5 pH 單位以內,蛋白質會開始溶離出來。 (3) 所用緩衝液的離子濃度,在不影響蛋白質與介質的結合能力下,儘量採稍高的濃度,以降低非必要性的吸附,通常在 10~100 mM (NaCl) 之間。 c. 膠体 pH 要先平衡好: 決定緩衝液的 pH 與離子濃度後,交換介質要先平衡在此緩衝液中,如膠体過濾法一樣,可在玻璃漏斗中進行。為加速平衡達成,交換介質可先用 10× 濃度緩衝液浸泡沖洗,然後再用 1× 者澈底清洗替換。 d. 管柱系統: 離子交換法所用的管柱系統,其要求是比膠体過濾法嚴格,最好使用附有 adaptor 的 Pharmacia 管柱,可降低 無效空間,避免梯度破壞。與膠体過濾相反,多使用矮胖型的管柱,太長並無必要。 e. 膠体裝填: 裝填方法與膠体過濾法一樣,但裝填要求反較不嚴格;裝填完成後,要以緩衝液洗過數個体積後方可使用。不能使用 Blue Dextran,用手電筒檢查有無氣泡。Sepharose 或 Sephacel 介質可耐高流速的壓力,但流速過快可能影響解析力。 2.3.4 管柱操作方法: a. 樣本蛋白質液: 樣本必須先溶在管柱所使用的緩衝液中,否則要先對緩衝液透析。樣本溶液的体積並無限制,因為蛋白質會結合到交換介質上,溶離下來時有濃縮效果;若目標蛋白質不 會吸附到介質,而直接通過交換介質,則其條件同膠体過濾法。 b. 溶離方法: (1) 如圖 2.10 所示可用 pH 或 鹽 梯度,溶離方式有 連續梯度 (continuous gradient) 及 階段梯度 (stepwise gradient)。 (2) 但 pH 的連續梯度不容易拉得好 (why?),因此一般較少使用,可改用階段梯度或 色層焦集法 (2.3.5),後者會在管柱內形成連續的 pH 梯度。 (3) 由於是以濃度梯度來溶離,每次加入管柱的緩衝液都不一樣,因此在離子交換管柱中,膠体上方不能積有緩衝液層 (無效空間,如圖 2.10 右),否則做好的梯度會在此處破壞,失去 pH 梯度的連續性,因此離子交換管柱最好能使用 adaptor。
c. 梯度体積: 每個梯度的体積會影響結果,通常溶離体積較大時,解析度較佳;但体積太大,會使溶離出來的蛋白質濃度變稀。而梯度的上下範圍 (如 0~0.3 M NaCl) 也要適當,範圍太寬或太窄,均會降低解析度,要以實驗試出最佳條件。 d. 膠体再生: (1) 蛋白質全部溶離出來後,交換介質要經過 再生 (regeneration) 完全後,才能再次使用。 (2) 以 1~2 M NaCl 即可洗去雜蛋白,澈底清洗可用 0.1 M NaOH 流洗;陰離子交換介質可用 1 M 醋酸鈉 (pH 2.0) 洗 1.5 個体積,再以緩衝液平衡完全,才能再度使用。 (3) 可測流出液的 pH 或離子濃度,是否與加入的緩衝液一樣。也可把膠体取出,在燒杯或漏斗中澈底清洗。大多數失敗是因於 再生不良! e. 批次法: 離子交換法不一定要在管柱中進行,也可在燒杯中以 批次法 (batch) 吸附並溶離蛋白質,一般應用在工業上的大量純化,其效果較差,但比較方便。 2.3.5 色層焦集法 (chromatofocusing): a. 也是一種離子交換法: (1) 若必須使用 pH 梯度進行溶離,則可改用 Pharmacia 發展的色層焦集法。 此法使用類似 DEAE-Sepharose 陰離子交換介質 (polyethyleneimine agarose),並在管柱中以特殊的緩衝液 (Polybuffer) 流洗以形成 pH 梯度。 (2) Polybuffer 含有如同 等電焦集法 (isoelectric focusing 是一種電泳方式) 所使用的 ampholyte,以較低的 pH 通入管柱,與介質上面的鹼性基團中和,由酸 (上方進入管柱處) 漸鹼 (出口處),直接在管柱膠体形成 pH 梯度。 b. 作用機制: 樣本蛋白質進入色層焦集管柱後,先遇到較高 pH 環境 (介質),通常高於其 pI 而帶負電,因此會結合到帶有正電的介質上。當 Polybuffer 開始注入管柱,降低環境 pH,使樣本分子失去負電荷而溶離下來;蛋白質便依 pI 大小順序,pI 高的先溶離出來,同時集中在其自身 pI 的地方,成為一條極細色帶,故稱為 焦集法。 c. 注意會產生沉澱: 有些酵素若處在其自身 pI 的環境,會發生不可逆的沉澱而失去活性,則不適用任何以 pI 為分離基礎的純化方法。因此色層焦集法通常較少使用,除非一定要以 pI 或 pH 梯度來作分離,否則儘量採用其他方法,目前已經很少看到。
|
|
|
▲ [Dowex] [DEAE] [CM]
|
|
|
|
|
| ▲ | |
| ▲ | |
| [Chromatofocusing] | |
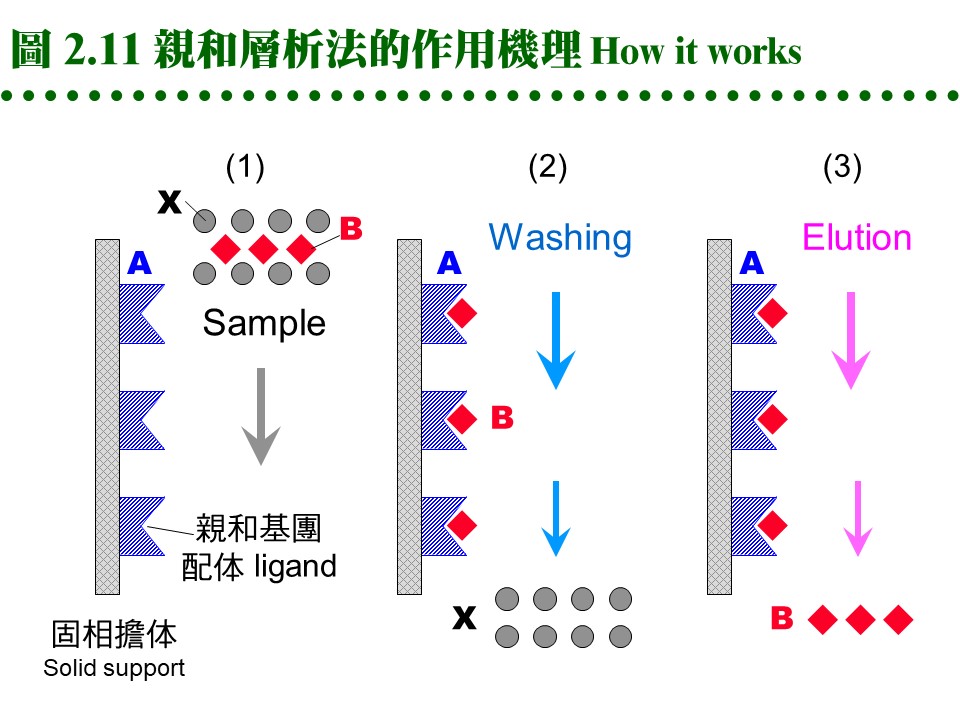
2.4 親和層析法:兩蛋白質分子間親和力之形成機制,請參閱生物化學 BCbasic 的專一性結合。 2.4.1 原理概述: 圖 2.11 說明純化過程之原理:
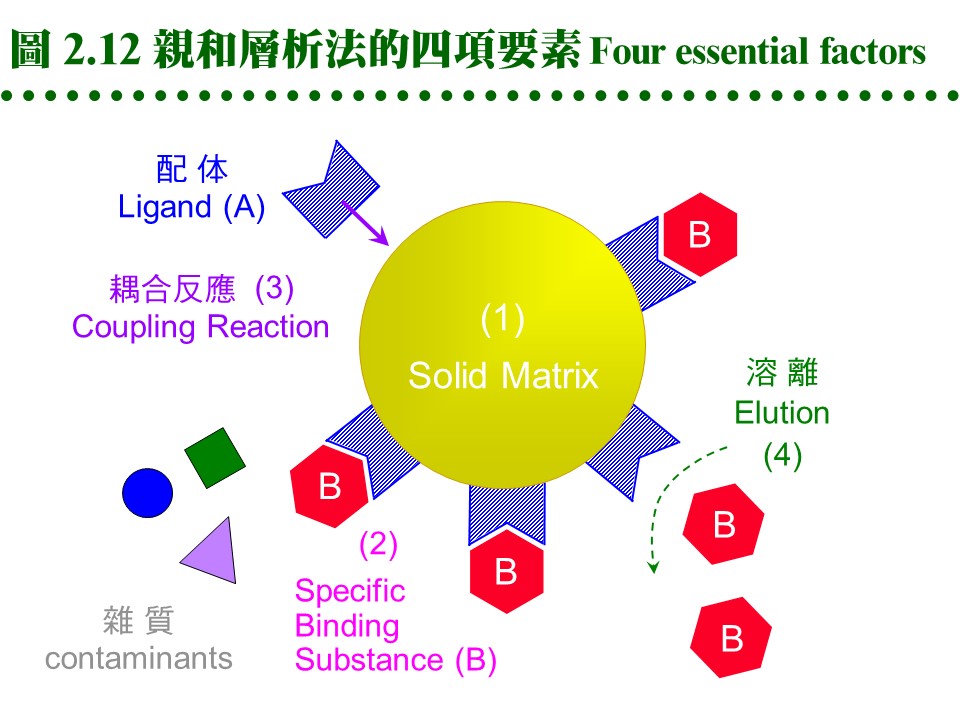
a. 固定相為親和基團: (1) 親和層析法的固定相為一固相擔体,上有 專一性親和基團 (A),流動相為溶離緩衝液。 (2) 當樣本通過管柱時,與親和基團有專一性的分子 (B) 吸附到固定相上,非專一性分子 (X) 則隨流動相洗出管柱。 (3) 留在定相上的分子 (B),可用酸或鹼溶離,或用專一性溶離分子溶離。 b. 親和法四項要素: 請參考圖 2.12:
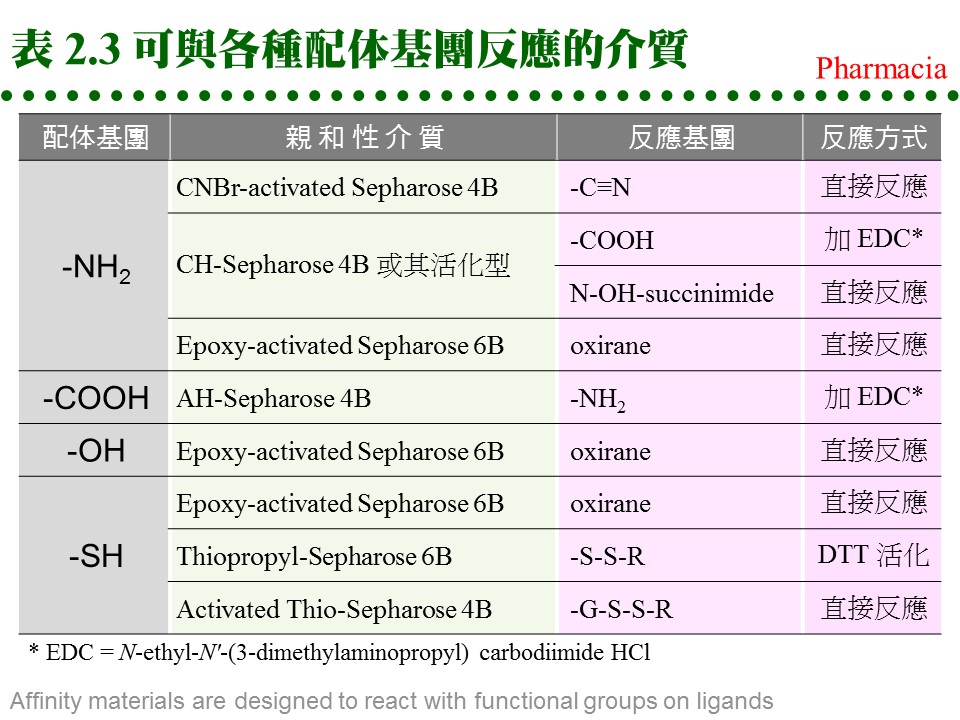
(1) 對所要純化的物質 (B),需有專一性 配体 ligand (A),而 A, B 之間要有專一性的親和力,其解離常數 (Kd) 約在10-4~10-8。 (2) 配体 (A) 可方便大量取得,且能經由耦合反應接到固相擔体,成為固定相。 (3) 擔体具有可與配体連結的基團,且 非專一性吸附力 低,通透性良好。 (4) A-B 結合成的 複合体 (complex),可以方便地解離,而不傷害 A 或 B。 2.4.2 親和吸著劑: a. 固相擔体: 材料種類很多,舉凡 洋菜糖 (agarsoe)、纖維素、玻璃砂、幾丁質、合成聚合物 均可使用;但用在蛋白質,仍以聚糖類為最佳。以 Sepharose 為例,可自行用 CNBr 活化,使糖分子接上 -O-C≡N (cyanate ester) 基,再與配体上的胺基反應。 b. 親和性介質:
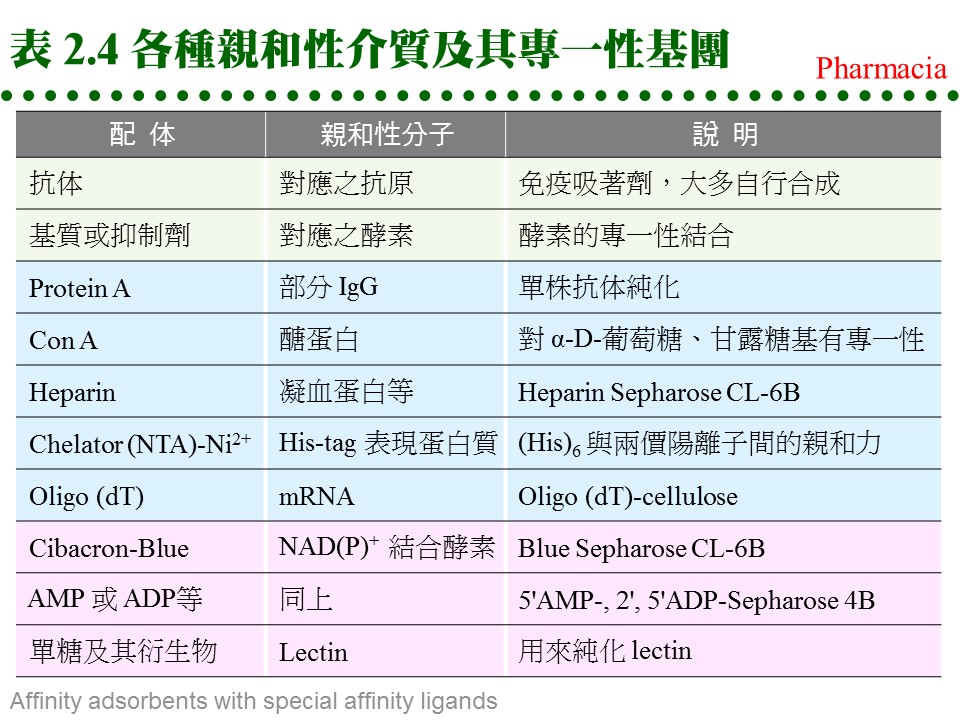
c. 共價層析法: 使用 Thio-Sepharose 時,樣本蛋白質是以 共價鍵 (雙硫鍵) 結合到親和介質上,然後再以 cysteine 或 mercaptoethanol 溶離下來。此法可以用來純化 papain 或如 urease 等含多個 -SH 基的蛋白質,特稱為 共價層析法 (covalent chromatography)。 d. 耦合反應 (coupling): (1) 介質與配体的 耦合反應 都相當簡便,介質先經緩衝液洗過後,加入配体溶液反應後,再加入填塞分子,除去介質上未完全反應的基團,裝入管柱流洗後即可使用。 (2) 注意耦合緩衝液及樣本液中,不能含有會競爭耦合反應的分子;例如使用 CNBr 活化的 Sepharose 時,不可用 Tris 或 glycine 緩衝液 (有-NH2 基)。 e. 注意 spacer arm: 有些親和層析法使用 spacer arm 來降低配体的立体障礙,但是 spacer arm 多為六到八碳的碳氫鏈,有相當強的非極性,若表現出疏水性層析的作用 (見下節),則可能對純化效果有正面或負面的影響。 f. 現成的親和吸著劑: 利用以上各種介質,可自行接上有用的配体,進行親和層析法,但商品也售有很多已經接好配体的成品,使用上更方便,例舉於表 2.4。
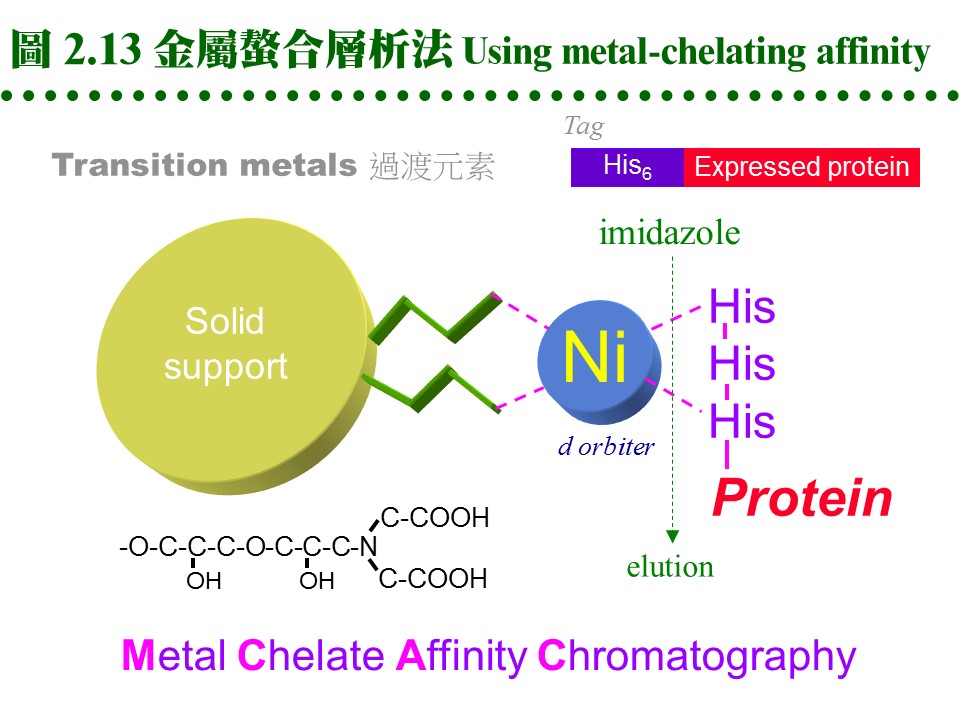
2.4.3 金屬螯合層析法: a. 許多蛋白質或酵素分子上帶有金屬離子,則此蛋白質可能會吸附該金屬。 b. 若把某金屬固定到固相擔体上,則此擔体會專一性地吸附需要此金屬的蛋白質。 c. 基因操作時,經常在表現蛋白質的端點,加上一段含有六個 His 的片段;則此表現蛋白質,可以吸附到含有鎳的吸著劑上,再以 imidazole 流洗出來 (圖 2.13)。 d. 這種擔体表面有可與金屬產生配位鍵的基團 (如 nitrilotriacetic acid, NTA),這些基團與金屬離子結合 (如鎳離子) 後,即可成為親和吸著劑。
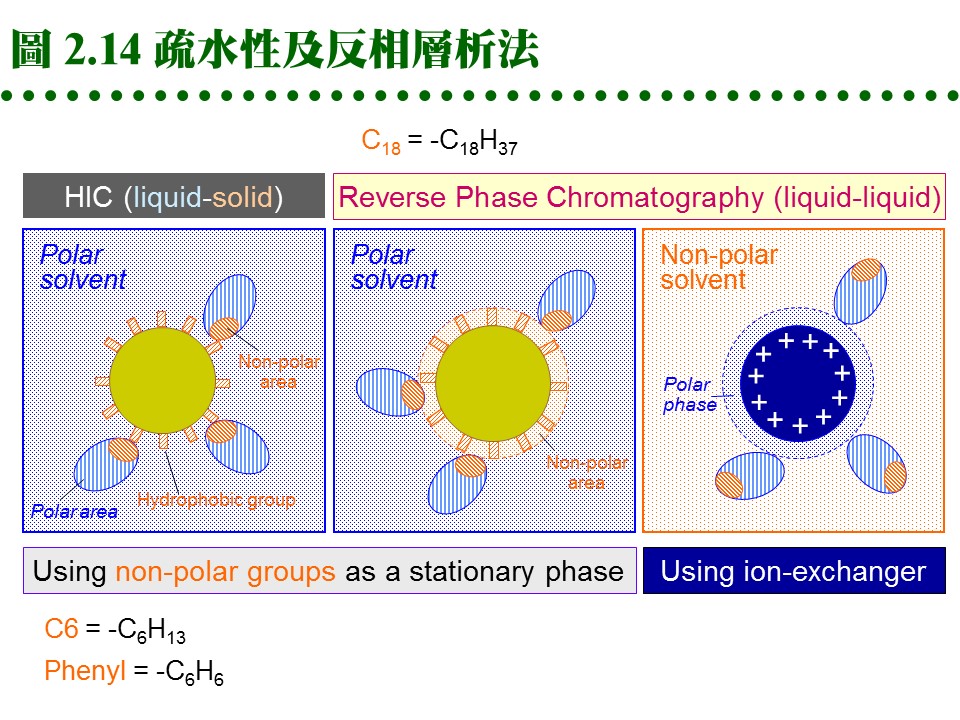
2.4.4 疏水性層析法: a. 作用機制: (1) 蛋白質分子表面有部份疏水性區域,若在一極性很強的環境中,則會被吸附在非極性的固定相擔体上,當環境的極性逐漸降低,就可被溶離出來,是為 疏水性層析法 (hydrophobic interaction chromatography, HIC)。 (2) HIC 沒有親和層析那麼強的專一性,較似離子交換法,但所根據的作用力,則是非極性基團間的疏水性引力。 b. 介質種類: (1) Pharmacia 有 Phenyl- 及 Octyl-Sepharose 兩種介質,後者疏水性較強。 (2) 通常樣本溶在 1 M 硫酸銨的緩衝液中通入膠体管柱,吸附上去的蛋白質,可提高緩衝液的疏水性來溶離,例如可用 ethylene glycol 之梯度溶離。 c. 反相層析法 (reversed phase): (1) 是 HIC 及離子交換法的綜合体,但屬於一種 partition 層析,可使用離子交換 (或類似 HIC) 的介質。 (2) 混合極性及非極性溶液為流動相,當流動相通入介質後,介質表面可固定其中的極性溶液 (若使用 HIC 介質則固定非極性者),樣本分子會在此二液相中進行 partition 分離。 (3) 因固定相及流動相的極性剛好相反,故名 reversed phase。請參考圖 2.14。
2.4.5 液相分配 (partitioning): a. 作用機制: (1) 在分析化學的純化方法中,使用分液漏斗在兩個液相間進行 partitioning 者,多用在有機小分子,因所用液相都是有機溶劑,蛋白質不易溶於其中。 (2) 若在水溶液中加入不同的親水性聚合物,造成密度的差異,則這兩個水溶液可分成兩相。若依蛋白質對此兩相之親和程度不同,而在此二相間進行分配 (partitioning) 則可達到分離效果。 (3) 可用的親水性聚合物有 polyethylene glycol (PEG), dextran, Ficoll 等。 b. 親和性分配法: 若在上述的聚合物分子上接有親和性基團,以吸引專一性的目標蛋白質,則稱為 親和性分配 (affinity partitioning)。
|
{BCbasic} |
| ▲ | |
|
[IMAC] |
|
|
|
|
|
[分液漏斗]
[PEG] |
|
2.5 HPLC 及 FPLCHPLC 為高效能液相層析法 (high performance liquid chromatography) 之意,也有說是 high pressure,因為要使用高壓推動溶離液,以加速層析過程。而 FPLC 則不用高壓,但流洗速度也很快 (fast performance),是因為所使用的介質通透性極佳之故。 這些都是屬於『硬体』的升級,也都可以應用在膠体過濾、離子交換、親和層析、反相層析等平台,而所根據的基本原理完全相同。 a. 解析力增加: 通常可降低介質的 粒子大小 以增加層析法的解析力,但是粒子太小造成流速太慢,反而使解析力下降。HPLC使用 silica 或樹脂等耐壓介質,以極細的粒子 (約 10 mm),在高壓下緊密裝填而成。使用時要在高壓下進行,因此速度比較快,通常在一小時左右可完成。 b. 使用方式廣泛: 較早 HPLC 都用於 adsorption 型式的層析法,例如以離子交換分離各種胺基酸等小分子。近來由於介質材料的發展,各種應用在大分子的液相層析法,均可用 HPLC 的方式來進行,不但加快分析速度,也使解析度提高很多。除了上述的離子交換法外,還可用在膠体過濾法、逆相層析法或親和層析法。HPLC 及 FPLC 系統,有可能完全取代傳統的低壓慢速液相層析法。 c. FPLC 系統: 是 Pharmacia 進一步發展的二代膠体介質與管柱体系,可以不用很高的壓力,在一小時內完成分離,而其樣本容量更大於 HPLC,可用在製備式純化上 (樣本量達 500 mg)。 |
[HPLC] |
|
[FPLC]
|
|
問題集 (每個問題不一定都有標準答案,甚至會引起很大的爭議,但這就是問題集之主要目的)1. 層析法為何一定要有兩相系統? (一相不行嗎?三相不是更好嗎?) [1] 2. 為何濾紙層析法 (PPC) 是一種 partition (液相-液相) 層析? [1] 3. 用 Sephadex G-25 脫鹽時,若因離子濃度改變,而致蛋白質發生沉澱,對實驗有何不利之處? [2] 4. 通常脫鹽的 Sephadex G-25 管柱均製成可丟棄式,這種消耗有必要嗎? [1] 5. 為何膠体過濾法是一種 partition 層析? [1] 6. 在進行離子交換法時,為何樹脂類的介質不適用在蛋白質樣本? [2] 7. 某蛋白質的 pI 是 4.2,若把緩衝液調到 8.5,則在此緩衝液下,應當使用那一種離子交換介質?請問在這樣的 pH 下進行離子交換層析,可能會有什麼問題? [3] 8. 為何 pH 的連續梯度不容易拉得好? [2] 9. 親和層析是 partition 或 adsorption 層析法?親和層析法為何需要使用固相擔体? [3] 10. 薄層層析 (TLC) 可兼有partition及adsorption兩種層析方式,各是如何操作的? [4] 11. 一隻直徑 2.6 cm 的管柱,長度 100 cm,想要裝填 Sepharose CL-6B 至八成的高度。 現有一瓶原裝的膠体,瓶子上面標明有膠体 750 mL,靜置後膠体沉降体積約佔四分之三的高度,其餘為上清液。你當如何取用膠体,剛好可以裝填所要的膠柱。 [4*] 12. 每年四、五月間,是研究生趕畢業的繁忙時段,冷房經常堆滿管柱,空間不敷使用。 有一研究生沒有找到位置,不得已只好在室溫跑膠体過濾,為了怕在室溫停留太久,又把流速增加 20%。結果出乎意料之外,出來的蛋白質峰都比原來在冷房裡跑的還集中,活性也稍稍增加。請檢討這個現象的可能原因。 [4*] 13. 某生以 Sephacryl S-100 膠体過濾法分離某蛋白質,在一般 PBS 緩衝液 (含 0.2 M NaCl)下流洗,通常此蛋白質都在大約 35 管分劃處溶離出來。但經春假放假三天回來,開動實驗重新裝填管柱,緩衝液也重新配製 PBS,卻發現蛋白質居然要在 60 管才會溶離出來,酵素總活性也降到三分之一。檢討他所用的管柱大小相同,所用的分劃收集器及分劃体積也都沒變,請問發生何事? [5*] 14. Hydroxylapatite 是何種物質?它是如何分離開單股與雙股 DNA? [3] 15. 有一次停電數小時之後,某生發現冷房內的管柱中,膠体都充滿了氣泡,請問是何原因?但他發現旁邊另一位同學的管柱膠体卻都沒有氣泡產生,又是為何? [4*] 16. 甲乙兩人同時用 DEAE 陰離子交換法純化酵素,甲生有最好的管柱 (有 adaptor)、梯度製造器、蠕動幫浦,而乙生除了有點錢買膠体、燒杯、緩衝液之外,什麼都沒有。經過一週後報告,乙生的結果卻比甲生好,活性及純度都較高,是如何辦到的? [5*] 17. HPLC 與 FPLC 系統有何異同?純化酵素時你會選擇何者? [2] 18. 進行離子交換法時,我們是採用較高的鹽濃度把蛋白質溶離下來,請說明為何高鹽濃度可以把吸附在膠体上的蛋白質洗下來? [3] 19. 進行離子交換法以鹽梯度溶離蛋白質時,若要分開兩個很相近的蛋白質峰時,有時會有下列現象發生,請說明為何: [5] a. 拉連續梯度不如階段 (stepwise) 梯度 b. 使用較長的膠柱不如較短的 c. 使用較寬的鹽梯度 (0~0.5 M) 不如較窄的 (0~0.2 M) d. 使用的溶離体積較小的 (如 150 mL × 2) 不如較大体積者 (如 200 mL × 2) 20. 血清蛋白質的 pI 大多在 7 以下,只有抗体 IgG 的 pI 為 8.3,請設計一離子交換程序,使用 DEAE-Sephacel,可以很快地純化 IgG。另外,IgG 的分子量約為 160,000,而另一類抗体 IgM 的分子量為其五倍多,請問你要用何種方法來純化 IgM? [4*] [題目後面方括號內的數字代表該題的難易程度,3 為中等而 5 最難回答,標有 * 為實際問題] |
|
| ▲ |