■ 分子消光係數
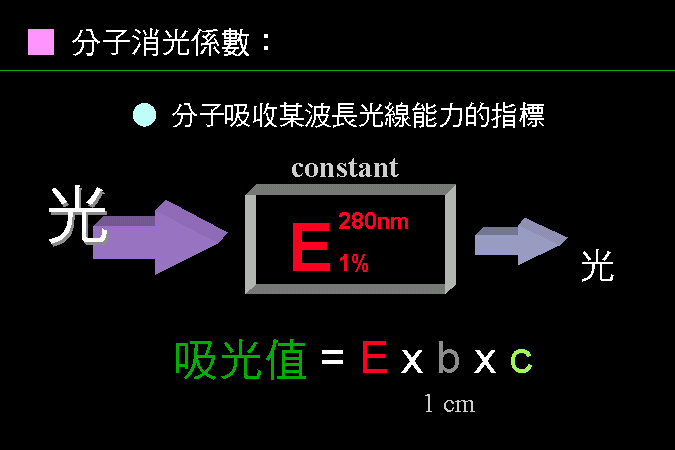
溶液中的分子會吸收光線中某波長的能量,吸光的多寡與溶液的濃度 c 有關。
純質分子的消光能力是其特定的性質,為一常數 E,不同的分子有不同的 E 值。
XA1300b
|
1 蛋白質定量法 |
2 酵素活性測定法 |
3 電泳檢定法 |
4 分子量決定法 |
5, 6, 7 其他工具 |
[AnaX] 到 [PurX]
補充說明用的幻燈片集成,看完該圖片後,請以『上一頁』或『回原主題』回到原處。
|
■ 分子消光係數 |
||
|
|
||
|
溶液中的分子會吸收光線中某波長的能量,吸光的多寡與溶液的濃度 c 有關。 |
||
|
純質分子的消光能力是其特定的性質,為一常數 E,不同的分子有不同的 E 值。 |
||
| ▲ 回原主題 ▲ TOP |
XA1300b |
|
|
|
||
|
蛋白質因為其芳香基團之故,在 280 nm 有吸光;因其骨架的 -C=O 基團,在 200 nm 有吸光。 |
||
|
會吸光的基團大多有電子的共振,其中最常見的是芳香族的基團,有 共軛雙鍵 (-C=C-C=C-) 的共振;每種蛋白質分子中的芳香基團含量不一,因此在 280 nm 的吸光能力就有很大差別,也因此各有不同的消光係數。 而 -C=O 基團是在 200 nm 附近有吸光,所有的蛋白質在此波長下,都有很強吸光,因為蛋白質骨架上的 -C=O 基團數量很多,而且不會因為蛋白質的胺基酸組成不同而有所差異。其精確度與準確度都很好,但因為 200 nm 波長的光線來源不穩定,而且會受到空氣中氧分子的干擾,因此較難應用。 |
||
| ▲ 回原主題 ▲ TOP |
XA1300c |
|
|
|
||
|
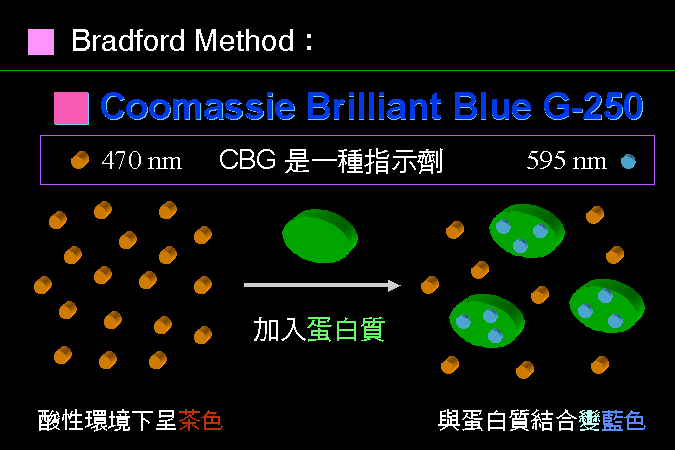
CBG 會吸附到蛋白質分子上,並且變成藍色。 |
||
|
CBG 有點像 指示劑,會在不同的酸鹼度下變色;在酸性下是茶色,在中性下為藍色。當 CBG 接到蛋白質上去的時候,因為蛋白質會提供 CBG 一個較為中性的環境,因此會變成藍色。當樣本中的蛋白質越多,吸到蛋白質上的 CBG 也多,藍色也會增強。因此,藍色的呈色強度,是與樣本中的蛋白質量成正比。 |
||
| ▲ 回原主題 ▲ TOP |
XA1400 |
|
|
■ 各種定量法的比較 |
||
|
|
||
|
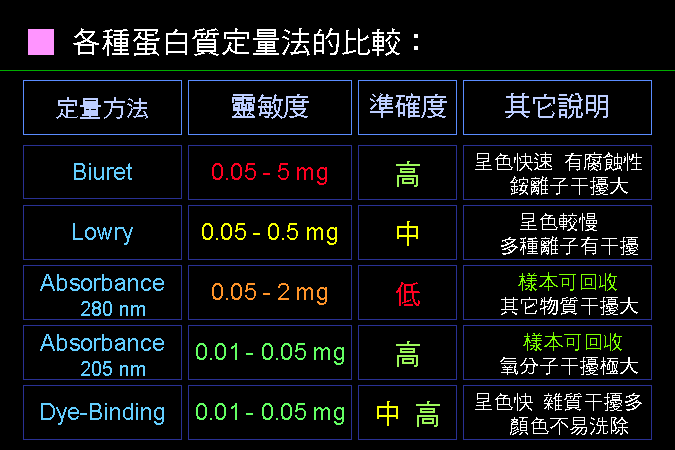
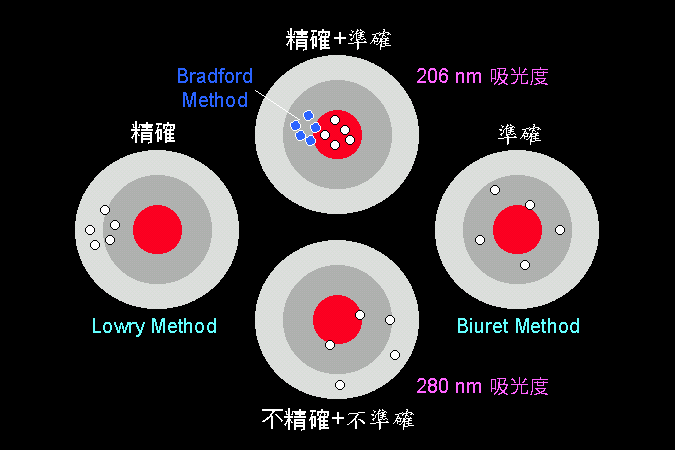
各種蛋白質的定量方法,各有其優缺點。 |
||
|
靈敏度就是 精確度,是看一種分析法能夠測到的最低量;最低限越低者,表示該分析法越靈敏。 準確度是看該分析方法準不準,會不會有假反應;不該測到的出來了,該測到的卻沒測到。高準確度如 Biuret 法,通常測到的都確實是蛋白質的量,不太會有假結果;低準確度者如 280 nm 吸光的方法,因為樣本蛋白質可能因為不含芳香族基團,而完全測不到。 再來比較 280 nm 與 205 nm 兩種吸光方法,後者既 精確 又 準確,前者 不精確 又 不準確。 因為,蛋白質在 205 nm 的吸光能力非常強,而且所有蛋白質的骨架上,都含有相同比例的 -C=O 基團,不會相差太多。 有關於精確度與準確度,請看下面打靶的例子。 |
||
| ▲ 回原主題 ▲ TOP |
XA1000a |
|
|
■ 精確度與準確度 |
||
|
|
||
|
精確度與準確度可以用打靶的例子來說明。 |
||
|
各種生化測量或分析方法,都有其應用上的評估標準,基本上是檢討其精確度與準確度;事實上與打靶的精準度很像。例如打 五發子彈,有些槍的精確度很好,五發都中在相當小的範圍內,但離靶心較遠,如左靶的 Lowry method;另有些槍雖然都中在靶心附近,但較散開,有準確度而缺精確度,例如 Biuret method 即是。 這是因為 Biuret 是根據蛋白質骨架上的 carbonyl (-C=O) 基團來定量,任何蛋白質的骨架都完全一樣,不會因蛋白質不同而有很大的差異,因此準確度較好。 而 Lowry 法是 Biuret 加上對 Tyr 的加強呈色,可以因此測得更微量的蛋白質;雖然其精確度是變大了,但並非所有的蛋白質都含有相同量的 Tyr,因此準確度變差。 同樣的,兩種以吸光度來測蛋白質量的方法,因為其所根據原理的不同,亦出現巨大的差異情形。由於 206 nm 測的也是蛋白質骨架上的 carbonyl 基團,因此非常準確;又由於其骨架上的 carbonyl 基團數目很多,蛋白質在 206 nm 的吸光非常之強,因此其精確度也很高。 而 280 nm 所測的是芳香基團胺基酸的吸光,每種蛋白質分子上芳香胺基酸的數目與種類都不一樣,因此不同的蛋白質的 280 nm 吸光就有極大的差別,測量起來因此不會太準。但我們可以先測得每個蛋白質的分子消光係數,以此來校正測量的吸光值,可求得正確的蛋白質含量。 |
||
| ▲ 回原主題 ▲ TOP |
XA1100 |
|
|
■ 酵素活性的測定 |
||
|
|
||
|
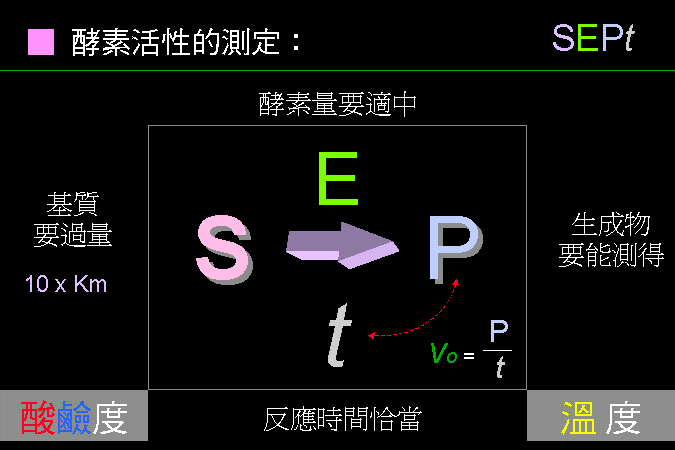
酵素活性測定的四個要點: SEPt |
||
|
當設計酵素的反應時,請考慮四個重要因素,可以用 SEPt 四個字母來代表: S (Substrate) 要使用大量基質,以便把反應推向右邊,通常是十倍的 Km。 E (Enzyme) 酵素量要適中,太高或太低都不好;但是通常酵素量都是待測值。 P (Product) 生成物要能夠方便偵測,否則要進行耦合反應,變成可測量的物質。 t (time) 反應的時間要恰當,並且固定下來;而酵素的反應速率 vo = P / t。 另外,要考慮反應溶液中的 pH 及溫度,不過這兩個因素通常也都固定。 |
||
| ▲ 回原主題 ▲ TOP |
XA2210a |
|
|
|
||
|
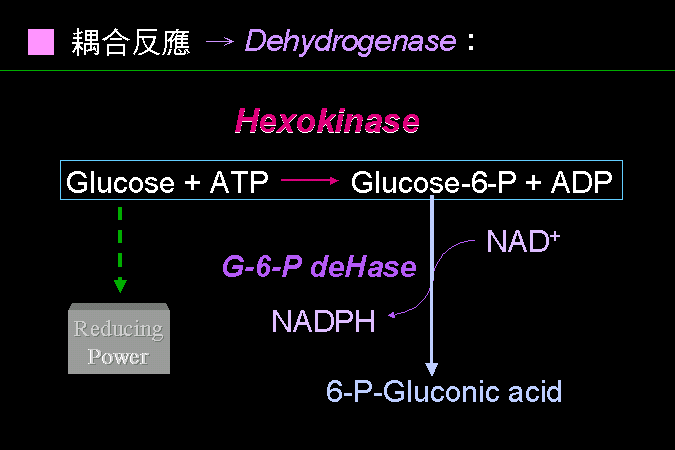
耦合反應可以有很多方式,能夠達到精確與準確者最好。 |
||
|
上面的 hexokinase,所得到的 Glc-6-P 無法直接以光度計偵測,因此要再以 Glc-6-P dehydrogenase 把 Glc-6-P 轉換成 6-P-gluconic acid,同時耦合著 NADH 的產生,NADH 的生成會造成 340 nm 吸光的變化,藉以偵測。 這個反應雖然也可以偵測基質葡萄糖的消失量,但通常都不這樣做。 因為酵素反應使用大量的基質,而要測得大量物質的小量變化,是較為不靈敏的方式。測量生成物的生成,是較為有利的;因為剛開始時,生成物的量為零,稍有一點點反應發生,即可靈敏地偵測到。 例如由 0 到 1 或 2 的變化,相當容易看到;而由 10000 變成 9999 甚至 9990,都不是極為明顯。 |
||
| ▲ 回原主題 ▲ TOP |
XA2210b |
|
|
|
||
|
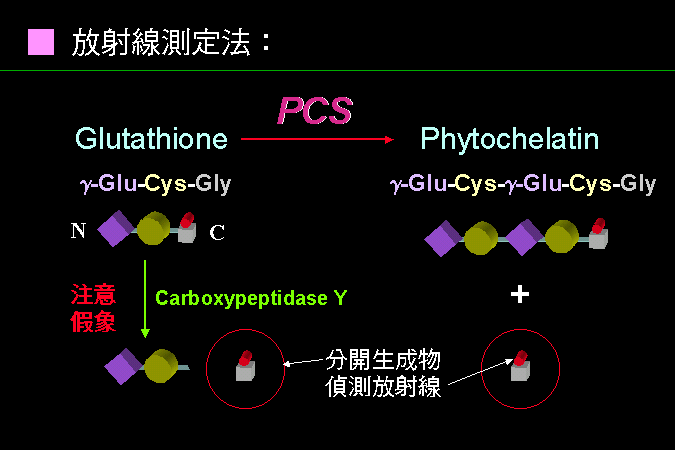
不得已才使用放射性基質,同時分離反應物與生成物也相當麻煩。 |
||
|
Phytochelain synthase (PCS) 不得不用放射性基質 (GSH),再測量生成物中有多少放射性 Gly;因為兩個 GSH 經 PCS 連接成 PC2 後,會放出一個 Gly 分子,而 GSH 的放射性是標示在 Gly 上面。麻煩的是,基質 GSH 與產物 PC2, Gly 都還混在一起,必須把放射性 GSH 與放射性 Gly 分開,通常都相當不易。 本案例可利用 GSH 與 Gly 在電荷性質上的差異,以離子交換介質吸附掉具有陰電性的 GSH。 更複雜的是,樣本中若有 carboxypeptidase,則會把 GSH 的 C-端 Gly 切下來,是一種假反應。 你能否以 PCS 為對象,設計一個更方便的活性分析方法? |
||
| ▲ 回原主題 ▲ TOP |
XA2210d |
|
|
■ SDS 如何作用 |
||
|
9 |
||
|
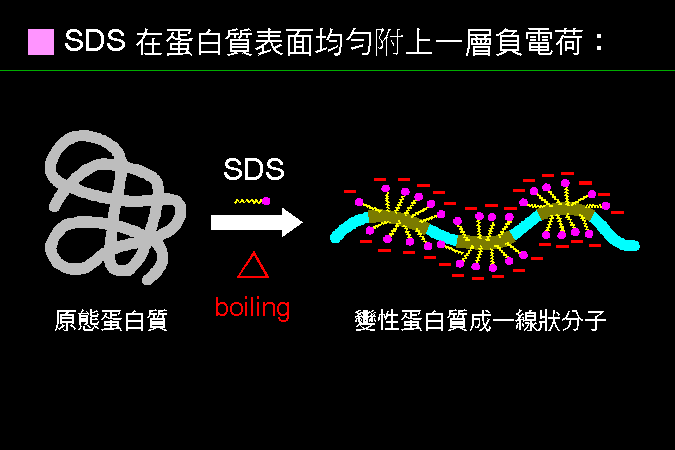
SDS 是一種界面活性劑,可以吸附到蛋白質的非極性區域。 |
||
|
原態蛋白質都有一定的分子構形 (左圖),若加入 SDS 並且加熱,則 SDS 分子上的非極性區 (黃色尾巴),會與蛋白質上的非極性區結合,並且使蛋白質變性,成為一線狀長條分子,上面佈滿 SDS 分子。SDS 分子的極性區 (洋紅色圓點),則露在外面,以增加親水性。這樣的蛋白質,幾乎不會有活性;但注意有些蛋白脢在 SDS 中還具有極佳的活性,如 protease K。 |
||
| ▲ 回原主題 ▲ TOP |
XA2220c |
|
|
|
||
|
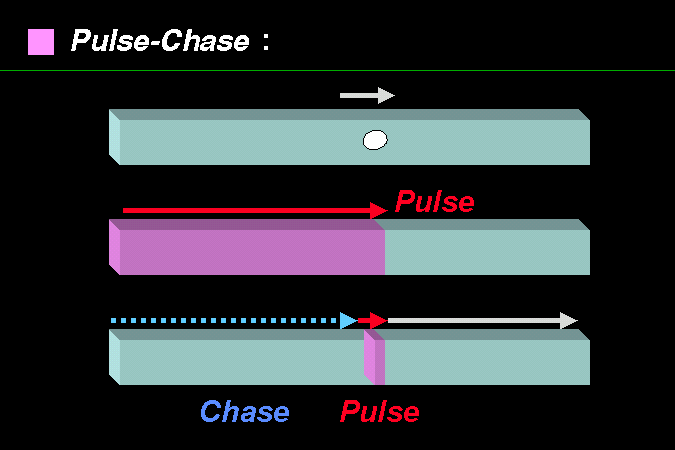
Pulse-chase 也是一種概念,可以應用在各種實驗的設計。 |
||
|
有一條透明水管,裡面有水在流,你如何能測得其流速? 若此時水管內出現一顆氣泡,看著氣泡的移動,你大概可以猜出流速;但可能不很保險,因為氣泡可能移動得較慢,也可能被卡住。若你在源頭處換到另外一種洋紅色的水,則看著洋紅色水柱的移動,你就可以準確地測量流速。事實上,洋紅色的水不須一直追加,只要有一點可以辨識位置的量,可再換回原來無色的水。把水換成洋紅色的動作,稱為 pulse;再換回無色的水稱為 chase。最常用的 pulse 是使用放射線物質。 很多實驗都可以用如此的概念來設計,以便觀察到進行中的現象,請不要忘記這個原則。 |
||
| ▲ 回原主題 ▲ TOP |
XA2200f |
|
|
|
||
|
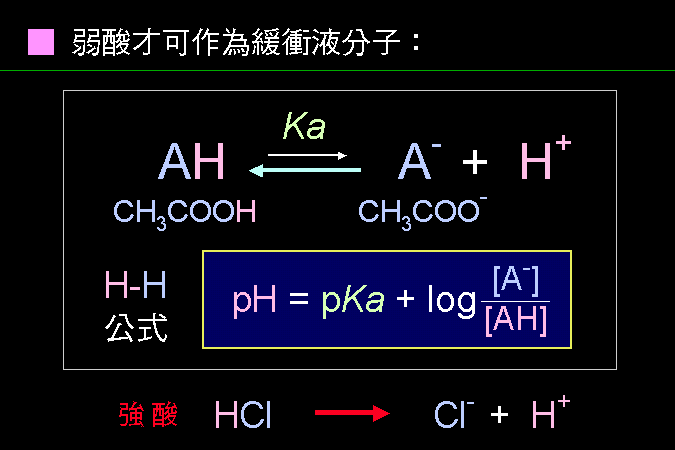
弱酸可以放出質子,也可以吸納質子,因此可以調節環境中的質子濃度。 |
||
|
這些可放出或吸收質子的基團,就是緩衝液的基本材料︰當環境質子太多,就趕快吸收質子;當環境質子太少,就釋放質子出來。 通常都是弱酸或弱鹼才可作為緩衝分子,因為強酸 (如 HCl) 只會釋出大量質子,不會回收;而強鹼只會拼命吸附質子,無法釋出。 Henderson-Hasselbalch (H-H) 公式可描述弱酸或弱鹼的緩衝行為。因為弱酸會釋出質子,也會吸附質子,因此其釋出與吸附間會形成一個平衡狀態,在平衡狀態下,可測得其平衡常數 (Ka);我們於是由弱酸的解離公式開始,由其平衡常數 (Ka) 依下頁的導法,求得 H-H 公式。 |
||
| ▲ 回原主題 ▲ TOP |
XA2310a |
|
|
|
||
|
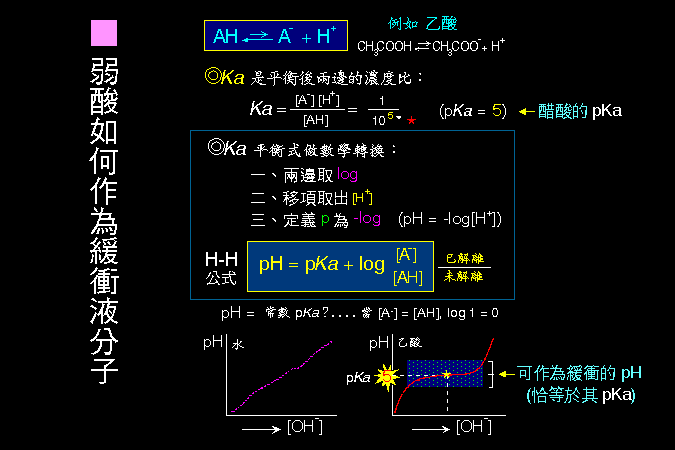
緩衝分子的作用,與其分子上基團的解離常數 (pKa) 有密切關係。 |
||
|
H-H 公式的 pH 即環境所測得的酸鹼度,pKa 為該緩衝分子的解離常數再取負指數;例如醋酸的 Ka = 10-5 (意指每十萬個醋酸分子中,有一個分子會解離,是弱酸的特性),則此 pKa = 5。 若環境的 pH 剛好 5,則依 H-H 公式︰5 = 5 + log([A-]/[AH]),右項 log([A-]/[AH]) 必須為零,而 log1 = 0,即 [A-]/[AH] = 1,即 [A-] = [AH]。這表示在 pH = 5 時,醋酸分子的組成中,有一半是在未解離的狀態 [AH],而另一半是已解離狀態 [A-]。在此情形下醋酸有最大的緩衝能力,因為不管外來的 pH 如何變化,都有足夠的 [A-] 或 [AH] 去吸附或釋出質子。以滴定法做實驗,也證實醋酸在 pH = 5 時,外加的酸或鹼都不易改變其整體酸鹼度。 因此,一種緩衝液的 緩衝範圍,與其 解離常數 有極大關係;pKa 等於 5 的弱酸,其緩衝範圍就在 4-6 之間。為何如此奇妙? 當然利用 H-H 公式可以上面的解釋說明之,但總合起來說,還是根基於解離常數的數學遊戲,以此來描述自然現象,沒有特別神奇的地方。 |
||
| ▲ 回原主題 ▲ TOP |
XA2310b |
|
|
|
||
|
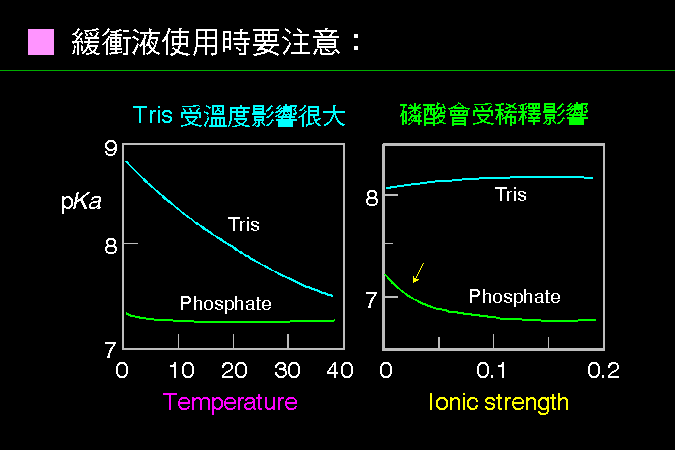
緩衝液的 pH 可能受到 溫度 與 濃度 的影響。 |
||
|
最常用的兩種緩衝液是 Tris 及磷酸,但都有一些應該注意的地方。 Tris 受到溫度的影響非常大,應該以將要使用的 pH 為調配標準,而且配 Tris 緩衝液要用特殊的電極。 磷酸緩衝液配成高濃度的 stock solution 時,要注意在低溫下很容易結晶出來,並且稀釋使用時 pH 會有點改變。 另外,很多實驗不能用某些緩衝液;例如親和吸著劑的耦合反應中,或者你的轉印色帶要切出來進行胺基酸定序,都不能用 Tris 或其他含有胺基的物質;澱粉磷解脢的活性分析中,因為是測量所釋出的磷酸,因此也不能用磷酸緩衝液。 使用每一種緩衝液,都要詳細研究它的特性與限制,以免影響實驗結果。 |
||
| ▲ 回原主題 ▲ TOP |
XA2310c |
|
|
|
||
|
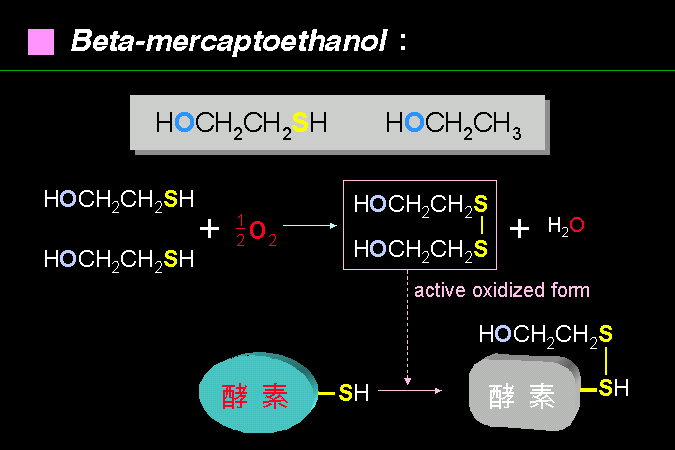
雖然可以幫助抗氧化作用,但要小心使用 b-mercaptoethanol。 |
||
|
b-Mercaptoethanol 的構造很像乙醇,但多了一個 -SH 基。此 -SH 基可以吸收氧分子,以防止氧化反應破壞蛋白質構形,可以說是犧牲了自己來拯救蛋白質。但它氧化後的物質,可能對蛋白質上的 -SH 基團產生修飾反應;若蛋白質的這個 -SH 基團對其活性很重要,則此蛋白質將反而被 mercaptoethanol 所害。 |
||
| ▲ 回原主題 ▲ TOP |
XA2310T1 |
|
|
■ DTT |
||
|
|
||
|
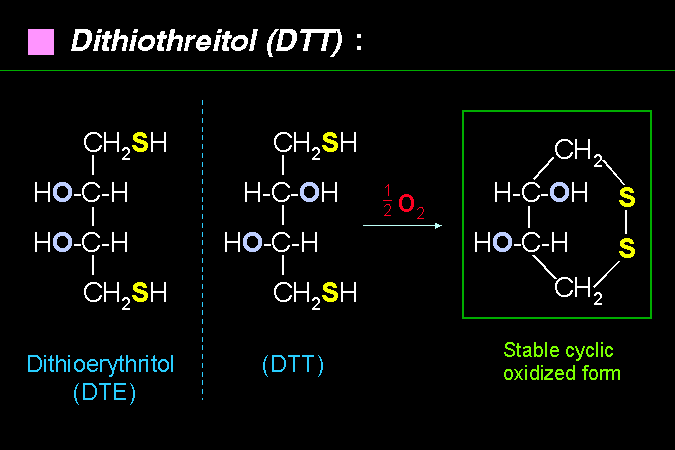
兩種可以抗氧化作用的添加物。 |
||
|
作用都類似 b-mercaptoethanol,而且不會有副作用,但價格較高。 |
||
| ▲ 回原主題 ▲ TOP |
XA2310T2 |
|
|
■ 細胞內外酵素分佈 |
||
|
|
||
|
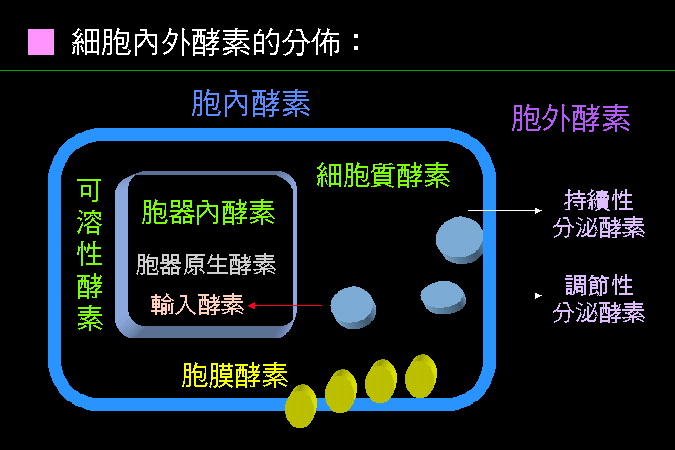
要考慮你的酵素,是屬於那一種蛋白質。 |
||
|
不同的蛋白質,會有不同的處理方法。 例如,胞膜上的酵素就相當麻煩,在安定性與活性分析上,就有相當的不同;其純化方法,也與水溶性酵素有很大差異。 |
||
| ▲ 回原主題 ▲ TOP |
XA2330a |
|
|
■ 各種蛋白脢 |
||
|
|
||
|
依酵素的 作用機制,蛋白脢可分為四大類,各有其特點。 |
||
|
凡是可以水解蛋白質上胜汰鍵的酵素,均統稱為蛋白脢。蛋白脢的種類非常多,我們大致歸納為四大類,均依其催化特性來命名。 例如 metal protease 是因為分子中含一金屬離子,此金屬離子不但可維持酵素的正確分子構形,也可以參與催化反應;Ser 及 Cys protease 是因為催化區上含有一個 Ser 或 Cys 胺基酸為主要的催化機團;而 Asp protease 也是因為分子上需要有兩個 Asp 基團,以便抓住水解所需的水分子。 每一類蛋白脢家族內,其成員的催化機制都相同,但催化目標的專一性不同;例如 Ser 家族內的 trypsin 嗜好水解鹼性胺基酸,而 chymotrypsin 喜歡較大的芳香基團。 |
||
| ▲ 回原主題 ▲ TOP |
XA2330b3 |
|
|
■ 影響泳動率因素 |
||
|
|
||
|
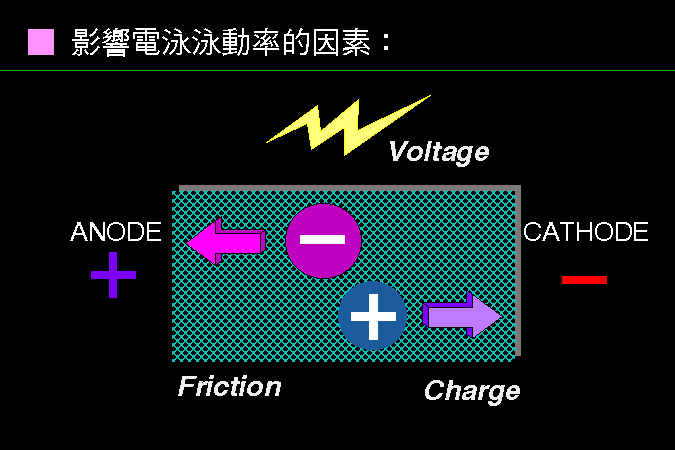
影響電泳膠片中,樣本泳動率的因素很多。 |
||
|
內在的影響因素,主要是樣本分子的『分子量、帶電性』兩種性質。 |
||
| ▲ 回原主題 ▲ TOP |
XA3110 |
|
|
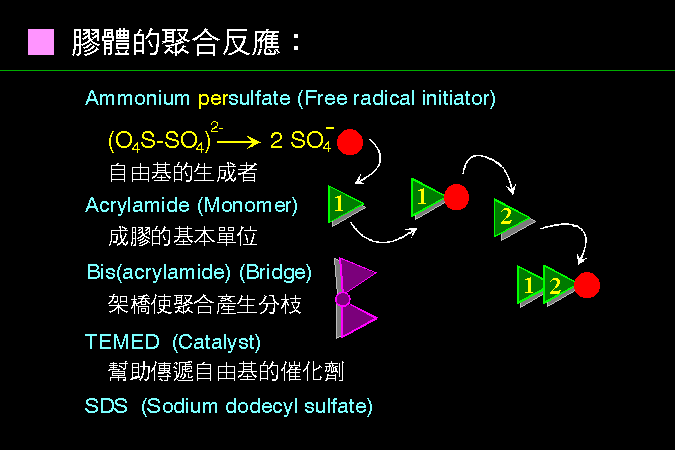
■ 膠體聚合反應 |
||
|
|
||
|
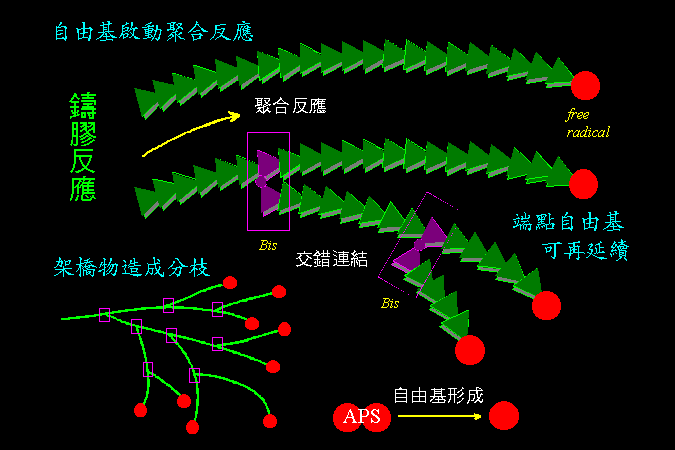
是一種 自由基 的 連鎖反應,把一個一個單元體連接成長鏈。 |
||
|
Ammonium persulfate (APS) 容易從分子中央裂解,成為對稱的兩半,平分原來的共價鍵,並形成 自由基 (上圖的紅色點代表自由基)。 此自由基開始攻擊一個 acrylamide 單元體,使後者成為自由基形式,再接續攻擊第二個單元體,因而串連起來;如此一直連接下去,即可成為長鏈巨分子。 Bis 是兩個 acrylamide 連在一起,若上述連鎖反應中含有少量 Bis,則有可能接到一個 Bis 分子,此長鏈將在此分叉,形成網狀構造 (請看下圖)。 TEMED 是催化劑,可以暫時保留自由基,以便充分供應連鎖反應所要的自由基。而有些電泳中,含有 SDS,即為 SDS-PAGE。 |
||
| ▲ 回原主題 ▲ TOP |
XA3222a |
|
|
|
||
|
膠體是一種立體的網狀構造,網目的大小會影響樣本的泳動率。 |
||
|
調配 acrylamide 及 Bis 的濃度,可以控制 膠體網目 的大小,也因此可以控制樣本分子的泳動率;以此來達到最佳的分離效果。 Acrylamide 的百分比較高者,其網目較小,可以分析較小的分子,通常解析力會較佳;但網目也不能太小,以免蛋白質的巨分子都跑不下來。 |
||
| ▲ 回原主題 ▲ TOP |
XA3222b |
|
|
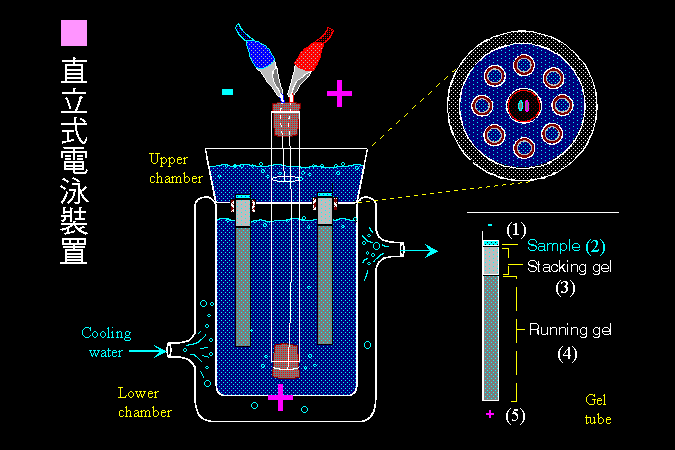
■ 古典的直立式電泳 |
||
|
|
||
|
農化系生化實驗課最早所使用的 直立式柱狀 電泳槽裝置。 |
||
|
雖然現在商品已經有很多方便的平板電泳設備,但若能親自體驗一次早期自製的垂直式柱狀電泳,則對電泳的發展,以及電泳原理的體會,將會有更深的認識。 |
||
| ▲ 回原主題 ▲ TOP |
XA3231b |
|
|
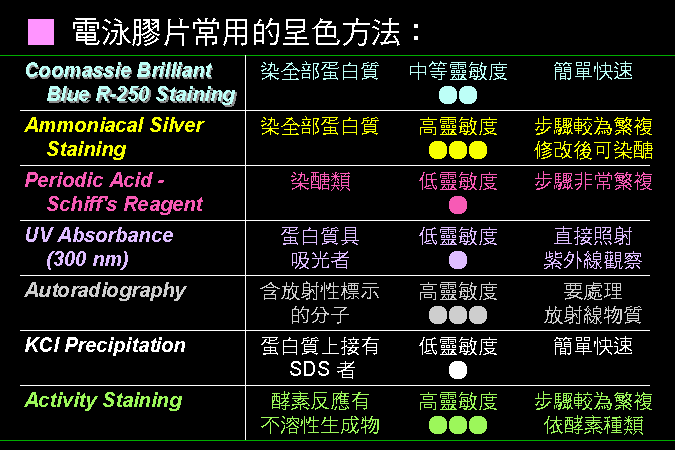
■ 常用的呈色方法 |
||
|
|
||
|
每種染色方法,都有其機制與原理,以及其特定的用途。 |
||
|
最常用的是 Coomassie Blue R-250 以及硝酸銀染色法,可染出一般蛋白質色帶;另外,酵素化學實驗以活性染色來染 澱粉磷解脢。 在細胞生物學的研究上,自動放射呈像法 autoradiography 極為常用。 |
||
| ▲ 回原主題 ▲ TOP |
XA3310a |
|
|
■ 結果不佳十大原因 |
||
|
|
||
|
電泳做不好,通常有很多原因,相當複雜。 |
||
|
像膠體完全不能凝結這種大問題,反而比較好解決。有時看起來一切似乎都很正常,只是結果不甚漂亮,例如膠片上色帶不集中,或是色帶有拖尾或瀰散,就較難以找出真正問題。 通常,問題都發生在各種 試劑的準備,要不是濃度不對,就是 pH 有問題,或者藥品已經失效 (APS, TEMED, Bis)。 若能確定所有的藥品都沒問題,用這些藥品所配製出來的各種溶液也沒問題,則應該已經是成功一半了。 |
||
| ▲ 回原主題 ▲ TOP |
XA3240a |
|
|
■ 二次元電泳 |
||
|
|
||
|
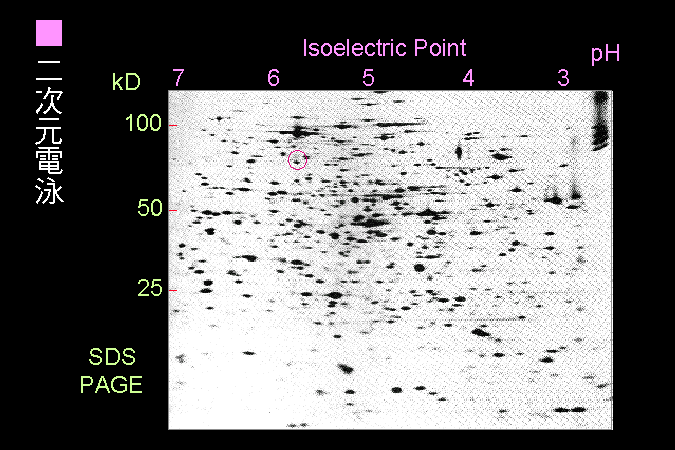
二次元電泳可以分析極為複雜的樣本。 |
||
|
最近興起的 proteome research,都想一下子把一個細胞的所有蛋白質全部分離開來,挑選其中有意義的色點,取出來直接進行分析其成分。因此,二次元電泳變得很重要,因為它的確可以達到分離的效果。通常第一次元都用等電焦集法,第二次元用 SDS-PAGE。要把二次元電泳跑好比較困難,至少平常的電泳要跑得很好才行。 |
||
| ▲ 回原主題 ▲ TOP |
XA3330 |
|
|
■ 免疫呈色機制 |
||
|
|
||
|
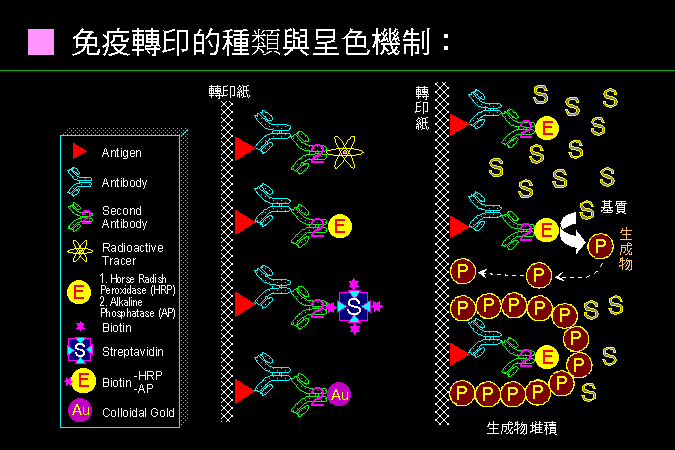
免疫染色的基理極為單純,但有很多不同應用方式。 |
||
|
當樣本蛋白質被印到轉印紙之後 (上面紅色三角),用其專一性抗體 (青色) 進行專一性辨認並結合,再用二次抗體 (綠色) 與標誌物的連結體標示 第一抗體;對這些標誌物進行檢定,如測量放射線、酵素呈色、電顯檢視等,就可以定位出抗原的位置。 上右圖是以酵素為例,加入基質被酵素催化產生有色的不溶性生成物,並且沈澱堆積在抗原色帶的地方。常用的標誌酵素是 horse radish peroxidase (HRP) 與 alkaline phosphatase (AP)。 這些標誌酵素也可以用 螢光呈色方法,可大大提高偵測靈敏度。 |
||
| ▲ 回原主題 ▲ TOP |
XA3340b |
|
|
|
||
|
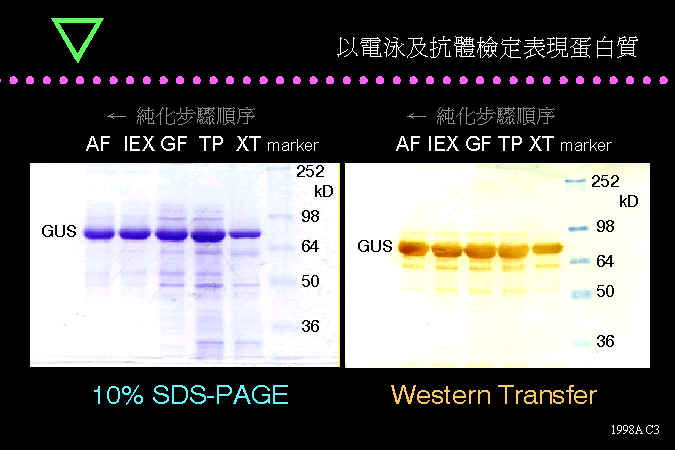
生物技術核心實驗 BCT 的蛋白質純化過程結果,並以免疫呈色追蹤目標酵素 GUS。 |
||
|
BCT 最後一個階段是把所表現的酵素 GUS 進行純化,分別進行粗抽取 (XT)、膠體過濾 (GF)、離子交換 (IEX)、親和層析法 (AF) 等。由左邊的 Coomassie Blue 染色圖可看到 GUS 越來越純;右圖的免疫染色,都只顯現出一條主要的 GUS 色帶。結果也顯示,GUS 的單體分子量約在 70 kD 左右。 |
||
| ▲ 回原主題 ▲ TOP |
XA4200b |
|
|
|
||
|
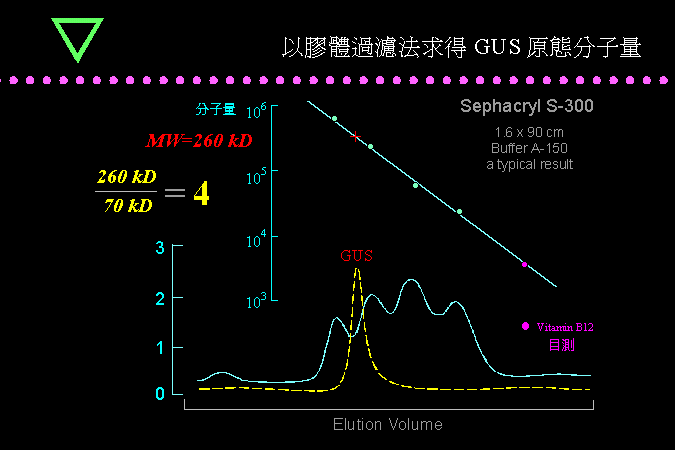
生物技術核心實驗 BCT 以膠體過濾定出目標酵素 GUS 的原態分子量 260 kD。 |
||
|
把 GUS 與標準分子量組合一起進行膠體過濾法,定出其溶離體積並與標準蛋白質比較後,內插可求出 GUS 的原態分子量為 260 kD。與上述單元體分子量 70 kD 比較,可推斷其四級構造為四元體。 |
||
| ▲ 回原主題 ▲ TOP |
XA4100a |
|
|
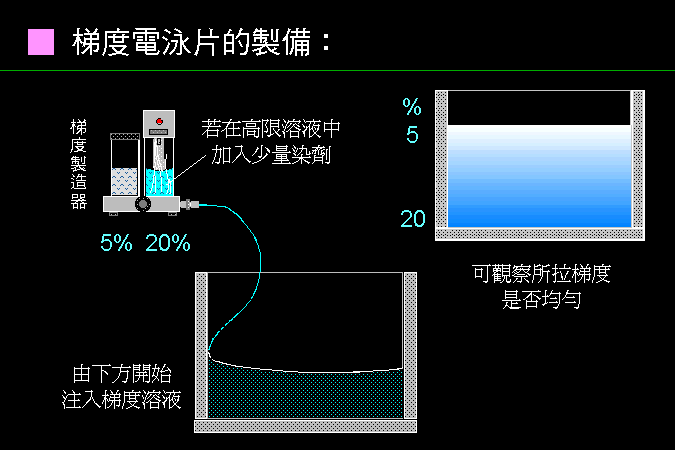
■ 製作梯度電泳膠片 |
||
|
|
||
|
梯度電泳膠片的製作相當困難,現在多有商品成品供應。 |
||
|
在實驗室自行製作梯度電泳膠片,需要相當的技巧與經驗,往往膠體還沒灌滿,就已經開始凝結;反之,把 APS 用量調很低,就遲遲不得凝結;即使能夠順利凝結,梯度也可能不甚均勻。 因此需要很多練習與嘗試失敗,才能有所成果。幸而現在商品多有已經預鑄好的梯度膠片,可省去很多時間與失敗經驗。但除非絕對必要,並不一定要用梯度電泳。 |
||
| ▲ 回原主題 ▲ TOP |
XA4200a |
|
|
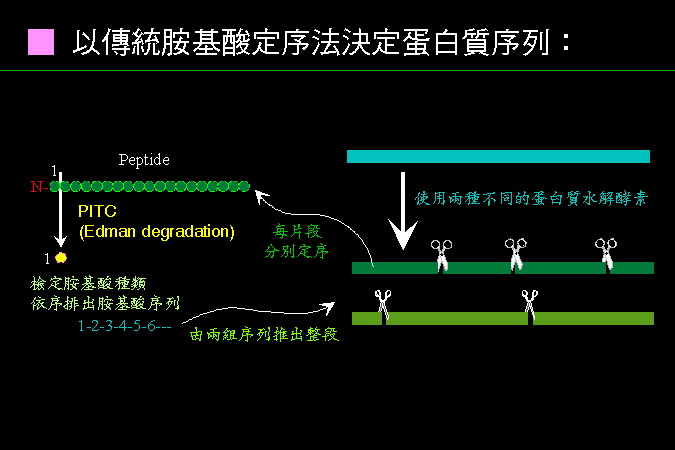
■ 胺基酸直接定序 |
||
|
|
||
|
傳統胺基酸定序要製備兩套不同切點的胜汰片段。 |
||
|
雖然可由 cDNA 推得蛋白質的胺基酸序列,但小規模的胺基酸定序仍有其應用與重要性。即使現在的自動定序儀,也還是用 Edman degradation 來定序,只是更加自動化,並且更靈敏、準確。 最近 proteome research 的發展,更要求快速檢定電泳後蛋白質色帶的胺基酸序列。除了傳統的 Edman degradation 外,也開始用 Mass 質譜儀 來進行定序。 |
||
| ▲ 回原主題 ▲ TOP |
XA5320d |
|
|
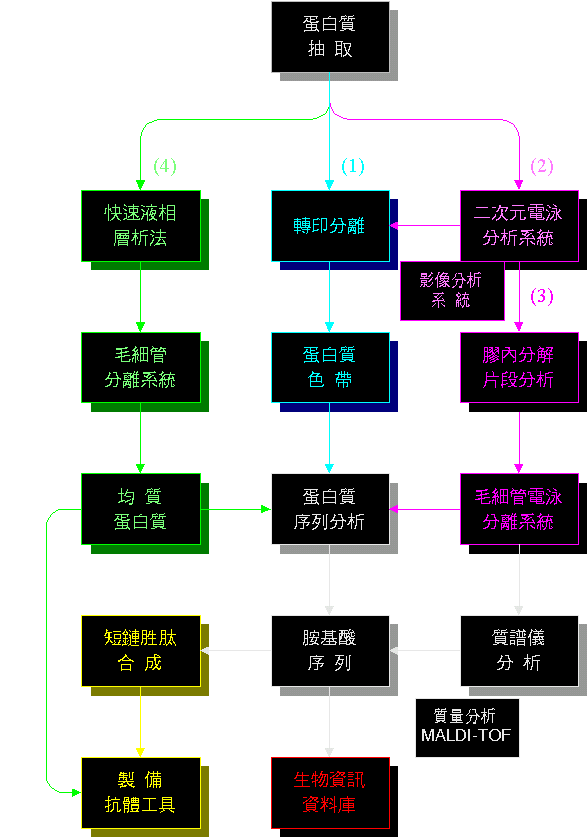
■ 蛋白質微量分析及檢定 (大圖) |
||
|
|
||
|
|
||
|
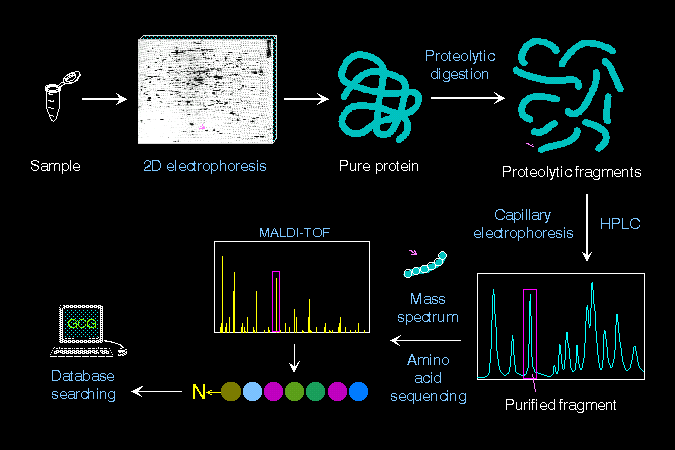
蛋白質得微量分離與分析系統,目的是快速得到指定蛋白質的身份。 |
||
|
可以把上面的各點,整理成以下四類工作: ● 微量純化系統: (1) 電泳及轉印 (2) 二次元電泳 (3) 膠體內水解 (4) 微量分離系統 ● 微量分析系統 ● 生物資訊系統 ● 抗體工具製備 |
||
| ▲ 回原主題 ▲ TOP |
XA7210 |
|
|
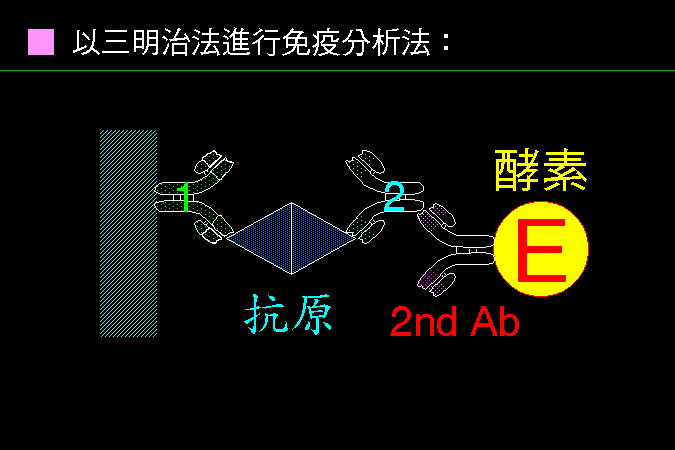
■ 三明治免疫分析法 |
||
|
|
||
|
這種三明治法也是一種常用的 ELISA 抗原檢定方式。 |
||
|
把抗體 (1) 固定在固相上,若加入的樣本中含有 抗原,則此 抗原 會被專一性地留在固相,同時洗去其它雜質;然後加入第二抗體 (2),此抗體也會與該 抗原 有專一性結合;接著用對抗第二抗體 (2) 的二次抗體-酵素連結體 (2nd Ab-Enz) 與抗體 (2) 結合,則可測定樣本中 抗原 的量。因為只有當樣本中含有 抗原 時,第二抗體與酵素的連結體才會留在固相上;因此測定固相上所留的酵素量,即可得知樣本中的 抗原 量 (呈正比)。 此法的專一性很高,因為樣本進行了兩次專一性辨認 (抗體 1 與抗體 2),因此背景值會很低。但是,樣本的 抗原 必須是 多價抗原,並可以誘生得兩種以上的抗體;另外,抗體 1 與抗體 2 必須來自不同動物來源,以免後來的 2nd Ab-Enz 連結體連接到抗體 1 上去。 當然,你可以直接在抗體 2 上面連接酵素,則不須 2nd Ab-Enz 連結體,但此種連結反應較難自己進行。 |
||
| ▲ 回原主題 ▲ TOP |
XA3340a |
|
|
■ 蛋白質的微量分析 |
||
|
|
||
|
科技的發達使得蛋白質的分析技術越來越靈敏,微量樣本即可得到所要結果。 |
||
|
微量 純化與分析技術,使得生化學家可以快速分離出一個蛋白質,並且準確判斷此蛋白質的身分。 通常從一個細胞樣本中,把總體蛋白質抽取出來,以二次元電泳分析之,定出所要找的目標蛋白質色點;切出此色點後,可直接在膠體中以專一性蛋白脢水解之,並以 HPLC 或毛細管電泳分離出單一胜汰片段,此一片段可用質譜儀或胺基酸定序儀分析其胺基酸序列,所得胺基酸序列由電腦資料庫中搜尋,即可判別原蛋白質的身分。 |
||
| ▲ 回原主題 ▲ TOP |
XA7220 |
|
|
■ 蛋白質體研究 |
||
|
|
||
|
由基因體所衍生出來的蛋白質體,是研究細胞生理與病理的重要工具。 |
||
|
Proteome 這個字是由 genome 衍生來的,故與 genome 有密切關係。Genome 是一個細胞染色體上全體基因的總稱,假設這些基因全數表現成蛋白質,此一總體蛋白質即稱為 proteome。 當各物種的 genome 一一被解出之後,我們可以翻出其總體蛋白質,由其所含的蛋白質種類,即可推測該細胞的代謝生理,或者其生理病變。要記得一個細胞內的總體基因,並不是每一個基因都正在表現,因此一個細胞的 genome 會因表現時期差異而有許多不同的 proteomes。 由於 genome 及 proteome 都是龐大的資料庫,如何應用電腦以及分析軟體,已成為一專門學科 bioinformatics,日益重要。 基因定序的工作目前幾乎已經完全 自動化,人體的全部基因在 2000 年中期應該可以完全解出來,而水稻的染色體序列,已經被 Monsanto 祕密完成;對於基因序列的使用,美國與英國已經呼籲應該由人類所共享。因此,我們應可跳過基因定序的浩大工程,直接開始思考『後基因時期』的新型態研究工作 (生醫報導 陳培哲:追求卓越計畫 - 基因體研究核心實驗室的策略)。 台灣是個小國,電腦科技已有相當的基礎,雖然軟體科技上並不十分成熟,但已經是我們最大的經濟籌碼;如何利用已知的基因序列,加上電腦科技,去找出一條可行的途徑,提升台灣在生物科技界的競爭力,則是我們能否在 21 世紀存活的關鍵。 |
||
| ▲ 回原主題 ▲ TOP ◆ 生物技術簡介 生物資訊學 連結 |
XA7310 |
|
[AnaX] 到 [PurX]
本網頁最近修訂日期: 2002/07/26
|
■ 各圖片主題 可連結回去原文主題 |
||
|
圖 例 |
||
|
Slide
|
||
|
圖片說明 |
||
|
詳細說明內文 |
||
| ▲ 回原主題 ▲ TOP |
XA |
|

 88
88