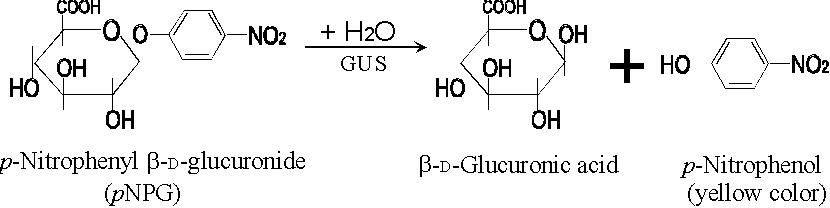
蛋白質定量分析
Bradford 的 dye-binding method 是利用 Coomassie brilliant blue G-250 (CBG) 可與蛋白質結合而 變色 的特性來定量 (Bradford, 1976);若試樣中的蛋白質量較多,則結合到蛋白質而變色的 CBG 也多,因而呈色較深。下例是以一組標準蛋白質為對象,製作一條蛋白質量與吸光度的標準校正線。
儀器用具:
ELISA 光度計 (Dynatech MRX) 可在一分鐘內測完一片滴定盤

Micro test tubes, 大頭針或噴火槍
藥品試劑:
(a) Buffer A-0
(b) BSA 標準品 (Bio-Rad, bovine serum albumin, 100 mg/mL)
(c) 未知濃度樣本 (unknown BSA, 0.8 mL, 由教師調配發給)
u以染料 Coomassie brilliant blue G-250 配製的溶液,勿與 膠體電泳染色 用的 CBR-250 混淆,各有其使用上的特色,不要互用。
標準品及樣本:
使用小試管準備一組 BSA (b) 標準品的系列稀釋:
|
Label |
BSA
(b) |
Buffer A-0 |
Final Conc. |
|
|
#5 |
500 mL |
0 mL |
100
mg/mL |
混合均勻 |
|
#4 |
400 mL |
100
mL |
80 mg/mL |
混合均勻 |
|
#3 |
300 mL |
200 mL |
60 mg/mL |
混合均勻 |
|
#2 |
200 mL |
300 mL |
40 mg/mL |
混合均勻 |
|
#1 |
100 mL |
400 mL |
20 mg/mL |
混合均勻 |
|
#0 |
0 mL |
500 mL |
0 mg/mL |
混合均勻 |
u配製一定要精準,否則校正直線不會很準確;各種試劑的添加,若 自稀往濃 取樣,則可以不用換吸管頭。
未知樣本:
再以上述 unknown BSA (c) 準備兩種未知樣本:
|
Label |
未知樣本
(c) |
Buffer
A-0 |
Final
Conc. |
|
X1 |
500
mL |
0 mL |
2× |
|
X2 |
250
mL |
250
mL |
1/2× |
一般樣本:
1) 通常由色析法所收集到的各分劃樣本,都可以直接進行蛋白質定量分析;但若其中含有某些干擾因子 (如含有 Triton ),或樣本的濃度太濃,都必須將樣本稀釋後再測。
2) 同一樣本的若干不同稀釋濃度,所測出來的最終蛋白質濃度會有差異,通常是以 稀釋度大者 較為準確。
方法步驟:
1) 每槽先加好 50 mL 標準品 (#0, #1 .... #5) 或未知樣本 (X1, X2),以 二重複 加入 96 孔滴定盤中,如圖 4.3 所示。
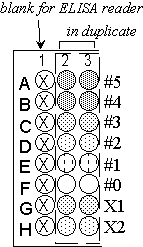
圖 4.3 微量滴定盤的使用
2) 每槽再加入200 mL Dye-Reagent (d),在全部樣本槽加完前,不要中斷添加;同時小心避免氣泡產生。
u本步驟是實驗成敗的關鍵!
3) 輕拍滴定盤一側,使添加的各成份均勻混合,注意 Dye-Reagent 密度較大,很容易沉積在下方而分層。 若還有小氣泡,小心以大頭針刺破,大量時可用噴火槍火燄燒破。
4) 靜置室溫 10 min。
5) 以 ELISA reader 測量 570 nm (或 595 nm) 的吸光值。
註解討論:
a. 在作圖紙上畫出標準品的校正線,並決定未知樣本的濃度 (如圖 4.4 )。
b. 請說明本分析方法的原理,並特別以蛋白質構造來說明。
c. 一般緩衝液或試劑中,何種添加物會影響 Bradford 法的呈色?
d. 請列出本分析方法的各操作步驟中,哪些會影響分析的準確度?
e. 還有哪些蛋白質的定量方法? 並請比較其優劣點。
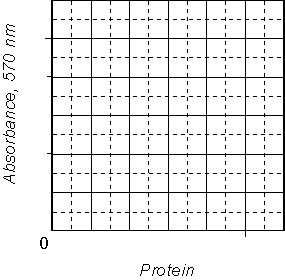
圖 4.4 繪製標準品校正線