Contents �D Chapter 0 �D Chapter 1 �D Chapter 2 �D Chapter 3 �D Chapter 4 �D Appendix
�x�W�j���ӯf�t �B �� ��
| �@ | �@ | �@ |
|
�ͪ��N��k ���@ |
�� �ͪ��N�֤߹��� BCT |
|
|
|
�@�^�ؿ� |
|
| �@ | �@ | �@ |
|
|
�ĤT�� | �@ |
| �@ |
Contents �D Chapter 0 �D Chapter 1 �D Chapter 2 �D Chapter 3 �D Chapter 4 �D Appendix |
�@ |
| �@ | �@ | �@ |
| �@ | �_�����X���R | |
| �@ | Northern Hybridization | �@ |
| �@ |
�x�W�j���ӯf�t �B �� �� |
�@ |
| �@ | �@ | �@ |
| �@ | �@ | �@ | |
| �@ | �@ | ||
| �@ | �@ | �� 3.1 | �@ |
| �@ |
3.1 RNA �s�� |
�@ |
�@ |
| �@ |
3.2 �H DIG �Щw�ֻı��w |
�@ | |
| �@ | �@ | ||
| �@ |
�@ |
�@ | |
| �@ |
|
||
| �@ | �@ | �@ | |
| �@ | �@ | |
| �@ |
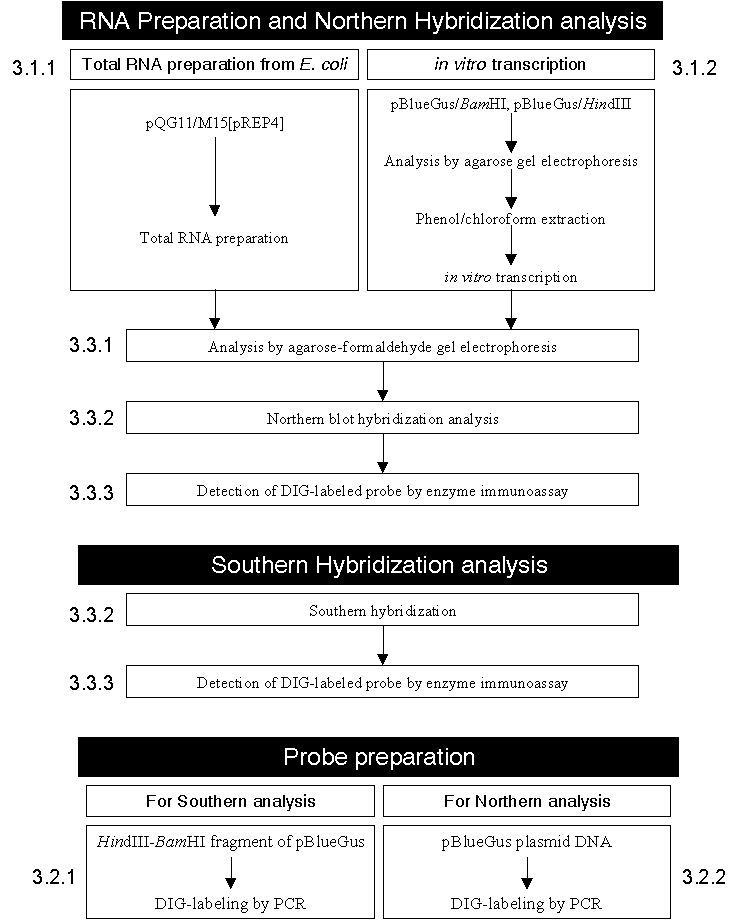
��]���{���Ĥ@�B�O�� RNA polymerase �H��������X���X mRNA�C �ѩ� mRNA �O�ӭM�����P��]����������A��ǦC�P�j�p���@�A�s�b�q�]���Y��ձ��A�@������ӭM������ RNA �� 1~5%�F��l RNA ���ݩ� rRNA (���� 80~85%) �� tRNA�A�b�u�ֲӭM�٦� small nuclear RNA (snRNA) �P small cytoplasmic RNA (scRNA) ���A���̤��O�H RNA �����������ѻP�ӭM���S�w�\�ध����A�]�A�J�ս���Ķ�P mRNA �ſ赥�C �]����������O��]���{�����n�ձ��I�A�b���R�S�w��]�����{���ήɡA�ե��o���F�Ѩ� mRNA ���ʽ�A�]�A���סB���c�Ϊ��{�q���h�赥�C �����n���Ъ� �_�����X���R (Northern hybridization analysis) �O�@��ΨӤ��R mRNA �̱`������k�F�b�i��_�����X�����ɡA���������s�� RNA�A�s�Ʀn�� RNA �H�ܩʬv�潦�� (denaturing agarose gel) �i��q�a���R��A�ߨ�H��Ӻ���L�k (capillary transfer) �Ψ�L��k��L���s���W�A�A�H�S�w�ֻı��w�i�����X���R�A�Y�i�����ؼ� mRNA �����פΦs�b�q�C �b�U������A�ڭ̤@�譱�N�H���� pBlueGus �i��M�~��������A�s�� GUS ��]�����N�� (sense) �P�ϷN�� (antisense) RNA�C�t�~�A�n�ۤj�z��� pQG11/M15[pREP4] �¤� RNA�A�H�K�H�_�����X�������R�b IPTG ���ɱ��p�U�A���{���� pQG11 ����a GUS ��]�����{���ΡC����y�{�p �� 3.1 �ҥܡC |
�@ |
| �@ | �@ | �@ |
| �@ | �@ | �@ |
| �@F3.1 |
| �@ |
|
|
| �@ |
|
�� 3.1 �_�����X���R����ާ@�y�{ (��j��) |
| �@ |
|
|
||
|
RNA �s�� |
||
| �@ | �@ | �@ |
| �@ |
�۲ӭM��� RNA ���B�J�D�n�]�A���}�ӭM�B�H�S�w��k�����ӭM�����J�ս�M DNA�B�αN RNA �I���X�ӡC �@��Ω}�ӭM����k�]�A�b�C�ŤU�H��ڬ�i�A�����Χ�����N�ӭM���}���A�ӱN�J�ս�P DNA ��������k�h�]�A�F�øѡB���������Ѩ��B�s��I���δ��䣱�����ߵ��C ���ױĥέ��@�ؤ�k�A�̻ݶO�ߪ̲��L�� RNase �����D�C �ѩ� RNase �|���� RNA�A�b RNA �s�ƹL�{���A��_�R���} RNase ���@�ΫK���F�M�w RNA �~��n�a������]�l�C �n�ѨM�o�Ӱ��D�A�����o�`�N���O�s�b��~�b���Ҫ� RNase�A�ȨD�Ҧ����羹�׳��O RNase-free�A�]���q�a�ѡB�����ζ콦���סB�����һݤ����G�ΤG�������A�����n�w���J�ӳB�z�A����ާ@�̤]�����H�ɱa�W��M (Sambrook et al., 1989)�C �t�@�Ӱ��D�h�O�ӭM���쥻�N���� RNase ���ʡF���ѨM�o���������D�A���b������ͪ����Ƥ���ߨ�i�� RNA ���s�Ƥu�@�C �Y���Ȯ��x�s�A�h���H�G�A��N�ͪ����ƫ�t�N���A�x�s�� -80�J �N���d�F���i�� RNA �s�ƮɡA�@�����X�x�s��N���d�����ơA���ߧY�H�G�A���i���覡���}�ӭM�A�þA�ɥ[�J RNA �Ѩ��w�IJG�A�H�קK RNase �@�ΡC �@�� RNA �Ѩ��w�IJG���t���S�w�� RNase ����A�Ҧp diethylpyrocarbonate (DEPC; Fedorcsak and Ehrenberg, 1966), guanidium thiocyanate (GTC; Chirgwin et al., 1979; McDonald et al., 1987) �� vanadyl-ribonucleoside complexes (Berger and Birkenmeier, 1979) ���A�H�K�b�ӭM�Q���}���b���A�Y�ɧ���ӭM�ت� RNase�C �ѩ�Y�� RNase ����|����S�w�������A�Ҧp in vitro translation ���A�b��� RNA �s�Ƥ�k�ɡA�����˲M���ҨϥΪ� RNase ����|���|�v�T���������A�H�K�\���@±�C |
�@ |
| �@ | �@ | �@ |
|
�ۤj�z��ߩ�� RNA |
�@ | |
| �@ | �@ | �@ |
| �@ |
���e�U�զ����w���O�c�v�X���{���� pQG11�ApQG11 �b JM109 �߮�w�i���{�X GUS�A�����{�q�ä����C �Y�n�O T5 promotor ���Ұʨ����n���ձ��A���i�@�B�N pQG11 ��Φܤj�z��ߵ߮� M15[pREP4]�C M15[pREP4] �i������{ lac repressor�ApQG11 �W T5 promoter �����ʱN�]�Ө������A���@���[�J IPTG �N��� GUS ��]�Q�j�q���{�C �b�o�@�`�������ءA�ڭ̱N���O�۸g IPTG ���ɻP���g���ɤ� pQG11/M15[pREP4] ������ RNA�A�H�K�H�_�����X�������R GUS mRNA �����{���ΡC �U���ұĥΪ���k�A�Ω�j�z��ߤΨ�L�����ʵ� RNA ������A����i��ɥ��H lysozyme ���ѲӭM���A�A�H sodium dodecyl sulfate (SDS) ���ѭ�ͽ轤 (Summers, 1970)�F����[�J�A�q����ƶu���G�N SDS�B�J�ս�Τj�z��ߪ��V���� DNA �@�֨I���U�ӡA�åH���ߪk�����A�s�d��W�M�G�� RNA �h�H�s��I���C ����L�{�ҨϥΪ� RNase ����� DEPC�C |
�@ |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
| �@ |
���ž_�����i�c 37�J |
�@ |
| �@ |
�B�D |
�@ |
| �@ |
�L�q���߾� |
�@ |
| �@ |
�������p (Hitachi U-1100 spectrophotometer) |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
�j�z��ߵ߮� pQG11/M15[pREP4] |
�@ |
| �@ |
LB ���i�G |
�@ |
| �@ |
Ampicillin (100 mg/mL) |
�@ |
| �@ |
Kanamycin (25 mg/mL) |
�@ |
| �@ |
IPTG (1 M) |
�@ |
| �@ |
Protoplasting buffer (15 mM Tris-HCl, pH 8.0; 0.45 M sucrose; 8 mM EDTA, stored at 4�J) |
�@ |
| �@ |
Lysozyme
(50 mg/mL) |
�@ |
| �@ |
Gram-negative lysing buffer (10 mM Tris-HCl, pH 8.0; 10 mM NaCl; 1 mM sodium citrate; 1.5% SDS) |
�@ |
| �@ |
Diethylpyrocarbonate
(DEPC) |
�@ |
| �@ |
���M��ƶu���G (�H 40 g ��ƶu���ѩ� 100 mL dH2O/DEPC) |
�@ |
| �@ |
����s��� 70% �s�� |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
�j�z��� pQG11/M15[pREP4] �����i�G |
�@ |
| �@ |
1) �N pQG11/M15[pREP4] ���ة� 2 mL �t ampicillin (100 mg/mL) �� kanamycin (25 mg/mL) �� LB ���i�G�A�� 37�J �_�����i�L�]�C |
�@ |
| �@ |
2) �j�馭��A�� 0.5 mL pQG11/M15[pREP4] �L�]���i�G�A�[�J 25 mL�t�� ampicillin (100 mg/mL) �P kanamycin (25 mg/mL) �� LB ���i�G�A�� 37�J �_�����i 2 h�C |
�@ |
| �@ |
3) �[�J25 mL 1 M IPTG�A�~���m�� 37�J �_�����i�C |
�@ |
| �@ |
4) �g�L 4 h ��A���X pQG11/M15[pREP4] ���i�G�A�}�l�i�� RNA �s�Ƥu�@�C |
�@ |
| �@ | �@ | �@ |
| �@ |
�s�� RNA�G |
�@ |
| �@ |
�� �`�N�I �i��H�U RNA �s�ƹ���ɡA���F�ϥΨ̤W�z�B�J�ҷdzƪ� pQG11/M15[pREP4] ���i�G�~�A�аO�o�V�U�л�����g IPTG ���ɤ� pQG11/M15[pREP4] ���i�G������ӲաC�C���l�������Q�ɡA�д��s���L�q�l���Y�H�K�y���ìV�C |
�@ |
| �@ |
1) ���X 4 �� 1.5 mL �L�q���ߺޡA���O�Хܬ� I-1, I-2, U-1 �P U-2�C |
�@ |
| �@ |
2) �� 3 mL �g IPTG ���ɤ� pQG11/M15[pREP4] ���i�G�������m��L�q���ߺ� I-1 �P I-2�C |
�@ |
| �@ |
3) �� 3 mL ���g IPTG ���ɤ� pQG11/M15[pREP4] ���i�G�������m��L�q���ߺ� U-1 �P U-2�C |
�@ |
| �@ |
4) �H 8,000 rpm (4�J) ���� 5 min�A�p�߭˱��W�M�G�A�åH�L�q�l�ɶq�l���ݯd���G��C |
�@ |
| �@ |
5) �U�[�J 0.5 mL protoplasting buffer�A�H�L�q�l�ޱN�j�z��ߥR���a�B�C |
�@ |
| �@ |
6) �A�[�J 1 mL protoplasting buffer �P 12 mL 50 mg/mL lysozyme�A�V�X�����A���m��B�D���@��15 min�C |
�@ |
| �@ |
7) �H 7,000 rpm �� 4�J ���� 5 min�C |
�@ |
| �@ |
8) �p�߭˥X�W�M�G�A�åH�L�q�l�ɶq�l���ݯd���G��C |
�@ |
| �@ |
9) �U�[�J 75 mL gram-negative lysing buffer �� 2 mL DEPC�A�H�L�q�l�ޤp���a�B�I����ީ����j�z��߭�ͽ���A�����Ƭ��C |
�@ |
| �@ |
�� �b�H�L�q�l���a�B�j�z�߭�ͽ���ɡA�@�� pellet �Q�R�������Y�������a�B�ʧ@�A�H�קK�j�z�� DNA �_���i��y�������D�C |
�@ |
| �@ |
�� DEPC �i��O�@�حP������A�ϥήɽЦb����o���ާ@�C |
�@ |
| �@ |
10) �N�L�q���ߺީ�J 37�J ���Ť��ѧ@�� 5 min�C |
�@ |
| �@ |
11) �@�Χ�������A�N���ߺ���B�D���R�m 5 min�C |
�@ |
| �@ |
12) �[�J 38 mL ���M��ƶu���G�A�H����Y���u���A�R���V�X���áC |
�@ |
| �@ |
13) �R�m��B�D���@�� 10 min�A���������զ�I���X�{�C |
�@ |
| �@ |
14) �H 13,000 rpm �� 4�J ���� 10 min�C |
�@ |
| �@ |
15) �N�W�M�G���ܷs���L�q���ߺޡA�å[�J 0.3 mL ����s��C |
�@ |
| �@ |
16) �V�X���ë�N���ߺީ�m�� -20�J �L�]�A�H�K�I�� RNA�C |
�@ |
| �@ |
�� RNA �b�s�뤺�۷�í�w�A�Y�������O�s�A�i���b�o�@�B�C |
�@ |
| �@ |
�j�ѡG |
�@ |
| �@ |
17) �H 14,000 rpm �� 4�J ���� 15 min�C |
�@ |
| �@ |
18) �p�߭˱��W�M�G��A�[�J 300 mL 70% �s��C |
�@ |
| �@ |
19) �H 14,000 rpm �� 4�J ���� 5 min ����A�p�߭˱��W�M�G�C |
�@ |
| �@ |
20) �N�L�q���ߺޭ˸m��ୱ�W 30 min�A�H�K���� RNA�C |
�@ |
| �@ |
21) �N RNA ���ѩ� 30 mL dH2O/DEPC�C |
�@ |
| �@ |
22) RNA�w�q�G�� 5 mL RNA�A�[�J 995 mL dH2O/DEPC �}������A�H�������p����b 260 nm �P 280 nm ���l���� (�O�o�ϥΥۭ^������)�A�íp�� RNA ���@�P A260/A280 ����ȡC |
�@ |
| �@ |
�� RNA ���G�b 260 nm ���l���Ȭ� 1.0 �ɡA�h�@�۷��� 40 mg/mL�A�Ө� A260/A280 ��������� 1.7~2.0 (DNA �h�� 1.8 ���k)�C |
�@ |
| �@ |
�� �o�|�� RNA �˥��N�������礤�A���O�H���Ǭv�潦��q�a�i����R (3.3.1.2)�C |
�@ |
| �@ |
���ѰQ�סG |
�@ |
| �@ |
a. RNA �s�ƹL�{�����n�[�J DEPC�H DEPC ���@�Τ覡����H ���| modify RNA �ܡH |
�@ |
| �@ |
b. �b�[�J���M��ƶu���G��A�ұo�쪺�զ�I���]�t���Ǧ����H |
�@ |
| �@ |
c. �H�W�z��k�ұo�쪺 RNA �I�����F RNA ���~�A�i���ٷ|�]�t�p�q DNA �P�J�ս�A�������P��z�Z�����n�i�檺�_�����X�����C |
|
| �@ | �@ |
�@ |
|
�M�~������� (in vitro transcription) |
�@ | |||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||
| �@ |
�ѩ� RNA �E�X�Q�@��ѼƭӦ��椸�զX�Ӧ��A�����n�b�պޤ����� RNA �E�X�Q��������ʦX�� RNA �ëD���ơA���o�Ӱ��D�b SP6 (Bulter and Chamberlin, 1982), T3 (Morris et al., 1986) �� T7 (Davanloo et al., 1984) �������餧 RNA�E�X�Q����Q�o�{�H��A���p�w�������[�C �o�D�n�O�]���o�� RNA �E�X�Q�Ѥ@���h�`�O (polypeptides) �Һc���A��b�i����������ɩҶ���{���Ұʤl (promoter) ���� 20 bp ���k�A�ӥB����_�l��m�D�`���T�A����į�}�n�A���F�ثe�i��M�~��������ɪ��̨ο�� (Krieg and Melton, 1984; Metlon et al., 1984; Studier and Moffatt, 1986; Tabor and Richardson, 1985)�C �n�i��M�~��������ɡA�ȻݱN�ؼа�]����� (subclone) �ܱa�� SP6, T3 �� T7 �Ұʤl���������A�Ϊ̹B�� PCR �N�b��]���@�ݥ[�W SP6, T3 �� T7 �Ұʤl���ǦC�A�Y�i�B�ι����� RNA �E�X�Q���ʦb�պޤ��X�����N�ѩΤϷN�� RNA�C �H���� DNA ���Ҫ��i��M�~��������e�A�����έ����Q�ۥؼа�]���@�ݤ��}�A�H�K�����������i��ܸ��骺�ǦC (run-on)�A���b��ܭ����Q��������קK�ϥΥؼа�]�]�a��������F�����Q�����n������ DNA �g phenol/chloroform �Ѩ�����A�Y�i�ΨӶi��M�~��������C �b�U�C�����ءA�ڭ̭n�ϥΪ�����O pBlueGus�C |
�@ | ||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||
|
���� DNA �øѤ����P�v�潦��q�a���R |
�@ | |||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||
| �@ |
������H BamHI �P HindIII �ø� pBlueGus�A�åH�v��q�a���R�����C |
�@ | ||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||
| �@ |
������G |
�@ | ||||||||||||||||||||||||
| �@ |
�L�q���߾� |
�@ | ||||||||||||||||||||||||
| �@ |
37�J ���Ť��� |
�@ | ||||||||||||||||||||||||
| �@ |
Mupid II �g�A�q�a�Ѥ�ű���� |
�@ | ||||||||||||||||||||||||
| �@ |
UV transilluminator |
�@ | ||||||||||||||||||||||||
| �@ |
���@���n |
�@ | ||||||||||||||||||||||||
| �@ |
��߱o�۾� |
�@ | ||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | ||||||||||||||||||||||||
| �@ |
���� pBlueGus DNA (�ѧU�д���) |
�@ | ||||||||||||||||||||||||
| �@ |
�����Q BamHI �P HindIII (BRL) |
�@ | ||||||||||||||||||||||||
| �@ |
10��Reaction buffer 2 (BRL) |
�@ | ||||||||||||||||||||||||
| �@ |
10��Reaction buffer 3 (BRL) |
�@ | ||||||||||||||||||||||||
| �@ |
Agarose |
�@ | ||||||||||||||||||||||||
| �@ |
1��TAE �q�a�w�IJG |
�@ | ||||||||||||||||||||||||
| �@ |
DNA ���l�q�аO (l DNA/HindIII, 0.5 mg/mL) |
�@ | ||||||||||||||||||||||||
| �@ |
10�Ѱl�ܬV�� (0.25% bromophenol blue; 0.25% xylene cyanol; 0.1 M EDTA; 50% glycerol) |
�@ | ||||||||||||||||||||||||
| �@ |
Ethidium bromide (500 mg/mL) |
�@ | ||||||||||||||||||||||||
| �@ |
��߱o 667 ���� |
�@ | ||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | ||||||||||||||||||||||||
| �@ |
1) ����� 1.5 mL �L�q���ߺޡA���O�Хܬ� #1 �P #2 ����A�̤U���ҦC���ǡA�v�@�[�J�øѤ����һݦU������ (��n��쬰 mL)�C |
�@ | ||||||||||||||||||||||||
| �@ |
�� �`�N�G�����Ҧ��������[�F����A�A�[�J�����Q�C |
�@ | ||||||||||||||||||||||||
| �@ |
|
�@ | ||||||||||||||||||||||||
| �@ |
2) �H����Y���u�L�q���ߺޡA�H�K���òV�X�ޤ��U������C |
�@ | ||||||||||||||||||||||||
| �@ |
3) ���Ƭ���A��m�� 37�J ���Ť��ѧ@�� 1~2 h�C |
�@ | ||||||||||||||||||||||||
| �@ |
4) �ЧQ�γo�@�q�ɶ��ѷӡy�ާ@�N�z�Ҵy�z���B�J�dzƤ@��1.2% �v�潦��C |
�@ | ||||||||||||||||||||||||
| �@ |
u Ethidium bromide �O�j�O�P������A�Y�T���ۤ�M��B�úN�I |
�@ | ||||||||||||||||||||||||
| �@ |
5) �۫��Ť��Ѩ��X�L�q���ߺޡA���Ƭ���Ȧs�B���ƥΡC |
�@ | ||||||||||||||||||||||||
| �@ |
6) ���i��q�a���R�ɡA�Ю��X 3 �� 1.5 mL �L�q���ߺޡA���O�Хܬ� #1, #2 �P #3�A�è̤U���ҥܦb�U���ߺv�@�[�J�A�q�� DNA �P 10�� �l�ܬV�� (��n��쬰 mL)�C |
�@ | ||||||||||||||||||||||||
| �@ |
|
�@ | ||||||||||||||||||||||||
| �@ |
7) �⽦�����ܹq�a�ѡA�`�J�A�q�q�a�w�IJG�A�H�L�q�l�� P20 �v�@�N�˥��l�J�`�X�Ʀ��ϲV�X���áA�äp�ߪ`�J�������˥��Ѥ��C |
�@ | ||||||||||||||||||||||||
| �@ |
8) ���n�q���u�H�w�q�� 100 V �i��q�a�A�ݰl�ܬV�����ʦܽ���T�����G�B�ɡA�����q���A�N���鲾�� UV transilluminator box�A�H��߱o�۾���� �C |
�@ | ||||||||||||||||||||||||
| �@ |
u DNA �a�ʤ�V�� (-) �V (+)�C ���~�A�b UV transilluminator box �W�����[�� DNA ��a�ɡA�S�����誺�P�������W�@����έ��n�A�H�קK UV �`�ˡF�t�� ethidium bromide ���v�潦��Х�ܱM�ήe�����I |
�@ | ||||||||||||||||||||||||
| �@ |
���ѰQ�סG |
�@ | ||||||||||||||||||||||||
| �@ |
a. ���˵� pBlueGus �� map�A�Q�Q����n��� BamHI �P HindIII�H �i�H��Ψ�L����ܡH |
�@ | ||||||||||||||||||||||||
| �@ |
b. �p�G�øѤ����������A�|����M�~��������y������v�T�H |
|||||||||||||||||||||||||
| �@ | �@ |
�@ |
|
�H phenol/chloroform �i�� DNA �Ѩ� |
�@ | ||||||||||||||||||||||||||||||||||
| �@ |
������G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�L�q���߾� |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Plasmid DNA �øѲ����G #1 (pBlueGus/HindIII), #2 (pBlueGus/BamHI) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Phenol/chloroform/isoamyl alcohol (25:24:1) �H�U²�٬� PCI |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Chloroform/isoamyl alcohol (24:1) �H�U²�٬� CI |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
3 M NaOAc (�L�Ķu pH 5.2) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�°s��� 70% �s�� |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u �`�N�I �i��o�@�������礧�e�A�Х��T�w pBlueGus/HindIII �P pBlueGus/ BamHI ��̪��øѤ������w�g�����C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
1) ��øѤ��� #1 �P #2 ���L�q���ߺޤ��O�[�J 182 mL �h���l���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
2) �H 200 mL PCI �Ѩ��@���A��Y�[�J PCI ��A�H vortex �_���V�X�A�æb�Ƿ����� 3 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
3) �H�L�q�l�� P200 �N�W�h�G���ܷs���L�q���ߺޡC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
4) �[�J����n�� CI�A�H votex �R���_���V�X�A�æb�Ƿ����� 3 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
5) �H�L�q�l�� P200 �N�W�h�G���ܷs���L�q���ߺޡC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
6) �[�J 0.1 ����n�� 3 M NaOAc (pH 5.2) �� 2.5 ����n����s��C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
7) �m�� -20�J �L�]�A�� -80�J ��m 30 min�A�H�K�H�� DNA�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | |||||||||||||||||||||||||||||||||
| �@ |
�j�ѡG |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
1) �ۧN���d���X�L�q���ߺޡA�H 14,000 rpm ���� 10 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u �`�N�G�ЧQ�γo�@�q�ɶ��ѷӡy�ާ@�N�z�Ҵy�z���B�J�dzƤ@�� 1.2% �v�潦��C Ethidium bromide �O�@�رj�O�P������A�Y�T���ۤ�M��B�úN�I |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
2) �p�ߦa�˥X�W�M�G�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
3) �}�}�[�J 0.5 mL �� 70% �s�� (�w����b -20�J �w�N)�C�p�ߤ��n�ʨ� DNA �H���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
4) �H 14,000 rpm �C������ 5 min ����A�˥h�W�M�G�A�ñN�L�q���ߺޭ˸m��@�i���b���åͯȤW 30 min�A�H�K���� DNA�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u �]�i�H�� SpeedVac ���� DNA�A�����p�߳B�z�A�H�K DNA pellet �����F�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
5) �H 10 mL dH2O/DEPC ���� DNA�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
6) �̷ӤU���A�� pBlueGus/BamHI �P pBlueGus/HindIII �U�� 1 mL DNA �H�v�潦��q�a�i����R�A�æ����@�סC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u DNA�@�פ�����ХH l DNA/HindIII �q���ѦҭȡC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
|
�@ | |||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | |||||||||||||||||||||||||||||||||
| �@ |
���ѰQ�סG |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
a. �Ҫ� DNA �O�_�����b�O�M�~���������_���\�����n�]�l�C |
||||||||||||||||||||||||||||||||||
| �@ | �@ |
�@ |
|||||||||||||||||||||||||||||||||
|
�M�~������� |
�@ | ||||||||||||||||||||||||||||||||||
| �@ |
������G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�L�q���߾� |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Plasmid DNA �øѲ��� pBlueGus/HindIII �� pBlueGus/BamHI |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
5��Transcription buffer (200 mM Tris-Cl, pH 8.0; 40 mM MgCl2; 10 mM spermidine-(HCl)3; 125 mM NaCl; BRL) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
NTPs (�t ATP, CTP, GTP��UTP �U 2.5 mM) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
RNase ��� RNasin (40 U/mL) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
T7 �P T3 RNA polymerase (BRL) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
dH2O/DEPC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
100 mM DTT (dithiothreitol) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
DNase (RNase-free) |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Phenol/chloroform/isoamyl alcohol (25:24:1), �H�U²�� PCI |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
Chloroform/isoamyl alcohol (24:1), �H�U²�� CI |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u �`�N�G�i��U�C����ɡA�аȥ�����M�I |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
1) �� 4 �� 1.5 mL �L�q���ߺޡA���O�Х� #1, #2, #3 �P #4�A�A�̤U���̧Ǧb #1 �P #2 �[�J�M�~��������һݦU���վ� (��� mL)�G |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
|
�@ | |||||||||||||||||||||||||||||||||
| �@ |
u �`�N�G�b�v�@�[�J�U�������վ��ɡA�L�q���ߺ�����ǷŧY�i�A�H�K�b�C�ŮɡADNA �J������w�IJG�ҧt spermidine ���ͨH���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
2) �H����Y���u���ߺ��ϲV�X���áA���Ƭ����m�� 37�J ���Ť��ѧ@�� 1 h�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
3) ���������ɡA�� #1 �P #2 ���O���X 4 mL�������A���L�q���ߺ� #3 �P #4 ���A�N #3 �P #4 �s��� -20�J �N���d���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
4) �� 1 mL DNase �[�� #1 �P #2�A�V�X���ë��m�� 37�J �@�� 15 min�A�H�K�h�� DNA �Ҫ��C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
5) ����������A�[�J 53 mL dH2O/DEPC�A�H 100 mL PCI �Ѩ��@���A�@�P�_����A���� 14,000 rpm 5 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
6) �H�L�q�l�� P200 �N�W�h�G���ܤ@�s���L�q���ߺޡA�U�h�� PCI �H 100 mL dH2O/DEPC �ϵѨ� (back-extraction) �@���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
7) �H�L�q�l�� P200 �N�W�h�G���ܨB�J 6) ���s���ߺޡC |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
8) ���p�⦸�W�h�G�E�����᪺�`��n�A�åH����n�� CI �Ѩ��@���A�@�P�_����A���� 14,000 rpm 5 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
9) �H�L�q�l�� P200 �N�W�h�G���ܤ@��s���L�q���ߺޡA�[�J 0.1 ����n�� 3 M NaOAc (pH 5.2) �� 2.5 ����n������s��C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
10) �V�X���áA�øm�� -20�J �L�]�A�H�K�H�� RNA�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
�j�ѡG |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
1) �ۧN���d���X�L�q���ߺޡA�H 14,000 rpm �� 4�J ���� 10 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
2) �J�Ӳ��h�W�M�G����A�[�J 0.5 mL 70% �s��C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
3) �H 14,000 rpm �� 4�J ���� 5 min�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
4) �h���W�M�G����A�N�L�q���ߺޭ˸m��@�i���b���åͯȤW�j�� 30 min�A�H�K���� RNA�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
5) �H 10 mL dH2O/DEPC ���� RNA �H���C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
���ѰQ�סG |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
a. RNasin �O�@�� RNase ��� (Blackburn et al., 1977)�A�@��i��M�~�������Ķ�����ɡA���ݲK�[ RNasin�A�H�קK RNA ���Ѫ����D�C |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
b. �H�W�z�M�~��������ҦX���X�Ӫ� #1 �P #2 RNA �U���h���H |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
c. �p��o�� #1 �P #2 RNA �U�� GUS ��]�����N�ѩΤϷN�� RNA�H |
�@ | |||||||||||||||||||||||||||||||||
| �@ |
d. �Y�n�H�W�z�M�~��������s�y�a cap �� mRNA�A�i�H���i��H |
�@ |
|||||||||||||||||||||||||||||||||
| �@ | �@ |
�@ |
�@�� TOP ��
|
|
||
|
�H DIG �Щw�ֻı��w |
||
| �@ | �@ | �@ |
| �@ |
DNA
���w�@��i�H�� 32P, 35S
�ΫD��g�ʪ���p biotin �� digoxigenin (DIG)
�[�H�Щw�F�Щw�ɧQ�� DNA �E�X�Q�����ʡA�b DNA
�E�X�������A�t�[�J [a-32P]
dATP, [a-32P] dCTP �� DIG-11-dUTP
���Щw����A�ҦX���X�Ӫ��Y�O�Q�Щw���ֻĤ��q�C
�ثe�`������k�]�A nick translation (Rigby et al., 1977), random
priming (Feinberg and Vogelstein, 1983) �P polymerase chain reaction (PCR;
Mullis et al., 1986; Saiki et al., 1988) ���C �Y���� DNA �i�� end
labeling �ɡA�i�H�i�� kinase �� filling-in reaction�C
�e�̥H [g-32P] ATP ��
phosphate donor�A�Q�� T4 polynucleotide kinase �����ʹ�a�� 5'-OH �� DNA �i��Щw�C�H�u�X������֥̻� (oligonucleotides)
5' �ݬ� -OH group�A�i�����Ω� kinase reaction�F�@�� DNA
���q�A�Y 5' �ݱa�� phosphate group�A�̦n���g phosphatase
�B�z�A�A�i�� kinase reaction�C Filling-in reaction �O�N DNA
�H�S�w�����Q���}�A�S�X recessed 3' termini ��A�Q��
Klenow �����ʧ� 3' �ݸɻ� (filling-in)�F
�]���A�u�n�[�J�A����
[a-32P] dNTP�A�Y�i�b DNA �� 3' �ݶi��Щw�C ���~�A�]�i�H���� bacteriophage T4 DNA
polymerase �� 3' |
�@ |
| �@ | �@ |
�@ |
|
Random priming |
�@ | |
| �@ | �@ | �@ |
| �@ |
DNA �E�X�Q�n�i�� DNA �E�X�����ɤ@�w�n�� �ޤl (primer)�A�]�����u��H�ޤl�Ҵ��Ѫ� 3'-OH ���_�l�I�i�橵�� (extension) ���u�@�A�Ӥ���N�a���s�X���s�� DNA (de novo synthesis)�C �@�����`�H�����X������֥̻Ĭ��ޤl�i�� DNA �E�X�����A�o�ɩT�M�i�H�ھڤw���ǦC�]�p�ֻĤޤl�A���]�i�H�ĥ� �{���ޤl (random primers)�C �ҿ׳{���ޤl�Y��ǦC���{���ǦC�A���g�S�O�]�p�A�ثe���γ̼s�̬O�H�۰ʤƾ����ҦX�����B����K�Ӯ֥̻Ī�����֥̻IJV�X���F�����i�椤�Y������@���{���ޤl���X��Ҫ� DNA�A�Y�i�� DNA �E�X�������i��C �H random priming ��k�i��ֻļЩw�A�O�H�{���ޤl�� Klenow fragment (large fragment of E. coli DNA polymerase I; Klenow and Henningsen, 1970) �i�� DNA �E�X�����A�æb�����L�{���K�[ [a-32P]dNTP �� DIG-11-dUTP ���Щw����C �Ӭ��F�קK random priming �]�o�ͩ���鳡���� DNA�A�̦n���n�����H���� DNA ���Ҫ��i��Щw�����A�����w���N�ؼ� DNA �۸�������X�ӡC |
�@ |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
| �@ |
�L�q���߾� |
�@ |
| �@ |
�m���D�ΦB�D |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
�Ҫ� DNA (pBlueGus �� BamHI-HindIII DNA ���q�A�N�ѧU�д���) |
�@ |
| �@ |
0.2 M EDTA, pH 8.0 |
�@ |
| �@ |
4 M LiCl |
�@ |
| �@ | �@ | |
| �@ |
Hexanucleotide mix (random primer) |
�@ |
| �@ |
dNTP mixture (1 mM dATP, 1 mM dCTP, 1 mM dGTP, 0.65 mM dTTP, 0.35 mM DIG-11-dUTP, pH 7.5) |
�@ |
| �@ |
Klenow enzyme (2 U/mL) |
�@ |
| �@ |
����s�� |
�@ |
| �@ |
70% �s�� |
�@ |
| �@ |
TE (10 mM Tris-Cl, pH 8.0; 1 mM EDTA) |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
u �H�U��������P�B�J�D�n�ѷ� random priming kit �s�y�t�өҴ��Ѥ����~�����ѡC |
�@ |
| �@ |
1) �� 100 ng �Ҫ� DNA�A�[�J�A�q�h���l���A����n�ɨ��� 15 mL�C |
�@ |
| �@ |
2) ��m�����[�� 10 min�C |
�@ |
| �@ |
u �аO�o�H���l�T�w�L�q���ߺޤ��\�����A�H�K���ߺަb�[���L�{���\�l�z�}�B�i���I |
�@ |
| �@ |
3) �[����A���t��L�q���ߺ��ܦB�D���A�R�m�Ƥ����C |
�@ |
| �@ |
4) ���Ƭ���A�̧ǥ[�J�U�C�T�ظվ��G |
�@ |
| �@ |
2 mL hexanucleotide mix |
�@ |
| �@ |
2 mL dNTP mixture |
�@ |
| �@ |
1 mL Klenow enzyme |
�@ |
| �@ |
5) �H���Y���u���ߺޡA�H�K�V�X���áC |
�@ |
| �@ |
6) �u�����߫�A�m�� 37�J ���Ť��Ѥ��@�� 2 h�C |
�@ |
| �@ |
u �����ɶ��ܤֶ��n 1 h�A�̪��i�����L�]�C |
�@ |
| �@ |
7) �@�Χ�������A�[�J 2 mL �� 0.2 M EDTA�A�H�K�פ� DNA �E�X�����C |
�@ |
| �@ |
8) �[�J 2.5 mL 4 M LiCl �� 75 mL �°s��A�V�X���áA�m -20�J �L�]�C |
�@ |
| �@ |
�j�ѡG |
�@ |
| �@ |
1) �� 13,000 rpm�A4�J ���� 15 min�C |
�@ |
| �@ |
2) �˥X�W�M�G�A�[�J 50 mL �� 70% �s��A�A���� 5 min�C |
�@ |
| �@ |
3) �˥X�W�M�G�í��� DNA�C |
�@ |
| �@ |
4) �̫�H 50 mL TE ���� DNA�C |
�@ |
| �@ |
���ѰQ�סG |
�@ |
| �@ |
a. �v�T�Щw�IJv���]�l�D�n�]�A�Ҫ� DNA ���«סB�ϥζq�P�����ɶ��F�b�i��Щw�����ɡA�Y DNA �ϥζq���� (�̰� 2 mg)�A�Τ����ɶ����� (�̪� 20 h)�A�i��o���Щw DNA �q�@�����h�C |
�@ |
| �@ |
b. �Щw����������H�s��I���Щw DNA�A�D�n�O���F�h�� unincorporated nucleotides�C �ѩ� unincorporated nucleotides �@�뤣�|�v�T����n�����X�����A�s��I���o�@�B�Υi�ٲ��C���p�G�һs�ƪ����w�O�n�Ω������X���� (in situ hybridization) �ɡA�̦n���i��s��I���C |
�@ |
| �@ |
c. ������H LiCl ���N�@��`�Ϊ��Q���i��s��I���A�O�]�� LiCl �b�s��̪����ѫפ���n�A�e���h���C |
�@ |
| �@ |
d. ���n�� DIG- �Щw�� DNA �i�� phenol/chloroform �Ѩ��A�]�� DIG �i��|�s�d�b organic phase�C |
|
| �@ | �@ |
�@ |
|
PCR |
�@ | ||||||||||||||||||||||||||||
| �@ | �@ | �@ | |||||||||||||||||||||||||||
| �@ |
PCR �O��~�ӷ����w�諸 DNA �W�ާN�A���μh���D�`�s�A�䤤�]�A�F�ֻı��w���s�ơC�p�G�@�q DNA �w�g�Q����ަܸ���W�A�n�H PCR �X�W�o�q DNA �ɡA�i�ھڽ��� multiple cloning sites ���䪺�ǦC�A��ξA�����ֻĤޤl�裡�X�ؼ� DNA�F����`�Ϊ��ޤl�]�A SP6, T3 �P T7 promoter primer, M13/pUC forward �P reverse primers ���C �i�� PCR �����ɭY�P�ɲK�[ [a-32P]dNTP �� DIG-11-dUTP�A�ҼW�ު� DNA �Y�Q 32P �� DIG �ҼЩw�A�ҥH PCR �O�s�Ʈֻı��w�D�`�K���n�Ϊ���k�C |
�@ | |||||||||||||||||||||||||||
| �@ | �@ | �@ | |||||||||||||||||||||||||||
| �@ |
������G |
�@ | |||||||||||||||||||||||||||
| �@ |
�L�q���߾� |
�@ | |||||||||||||||||||||||||||
| �@ |
PCR ������ |
�@ | |||||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | |||||||||||||||||||||||||||
| �@ |
�Ҫ� DNA�G pBlueGus ���� DNA (~50 pg) |
�@ | |||||||||||||||||||||||||||
| �@ |
�ֻĤޤl��G T3 promoter primer �P T7 promoter primer (�U 2 mM) |
�@ | |||||||||||||||||||||||||||
| �@ |
�q���o (�O�_�ݭn�� PCR �����������өw) |
�@ | |||||||||||||||||||||||||||
| �@ |
PCR DIG probe synthesis kit (Roche), ���t�G |
�@ | |||||||||||||||||||||||||||
| �@ |
Taq DNA polymerase (Expand High Fidelity, 3.5 U/mL) |
�@ | |||||||||||||||||||||||||||
| �@ |
10��PCR DIG probe synthesis mix (2 mM dATP, dCTP, dGTP, 1.9 mM dTTP, �� 0.1 mM DIG-11-dUTP, pH 7.0) |
�@ | |||||||||||||||||||||||||||
| �@ |
Expand high fidelity buffer with MgCl2 (10��) |
�@ | |||||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | |||||||||||||||||||||||||||
| �@ |
u �H�U��������D�n�ѷ� PCR DIG probe synthesis kit �s�y�t�өҴ��Ѥ����~�����C |
�@ | |||||||||||||||||||||||||||
| �@ |
1) ���@�� 0.5 mL �L�q���ߺޡA�̤U���v�@�[�J�U���վ� (��� mL)�G |
�@ | |||||||||||||||||||||||||||
| �@ |
|
�@ | |||||||||||||||||||||||||||
| �@ |
2) �H����Y���u���ϦU���վ��V�X���áC |
�@ | |||||||||||||||||||||||||||
| �@ |
3) �u�����߫�A�}�}�[�J100 mL �q���o�C |
�@ | |||||||||||||||||||||||||||
| �@ |
u ���� PCR ���������ݨϥ��q���o�C |
�@ | |||||||||||||||||||||||||||
| �@ |
4) �b PCR �������W�]�w PCR ��������G |
�@ | |||||||||||||||||||||||||||
| �@ |
Denaturation |
�@ | |||||||||||||||||||||||||||
| �@ |
�X�W�����G |
�@ | |||||||||||||||||||||||||||
| �@ |
30 �`���� [94�J/45 s ® 55�J/1 min ® 72�J/2 min] |
�@ | |||||||||||||||||||||||||||
| �@ |
�̫ᤧ elongation step�G 72�J/10 min |
�@ | |||||||||||||||||||||||||||
| �@ |
5) �N�L�q���ߺ��ܤ������W�A�ö}�l�i�� PCR �����C |
�@ | |||||||||||||||||||||||||||
| �@ |
6) ����������A�H�L�q�l�ާl�X�q���o�A�è��X 5 mL PCR ���������A�i��v�潦��q�a���R�C |
�@ | |||||||||||||||||||||||||||
| �@ |
���ѰQ�סG |
�@ | |||||||||||||||||||||||||||
| �@ |
a. PCR �Щw�����ҧe�{�� molecular mass ���Щw�̤j�C |
�@ | |||||||||||||||||||||||||||
| �@ |
b. �v�T PCR �������]�l�D�n�]�A���ǡH |
�@ | |||||||||||||||||||||||||||
| �@ |
c. �H PCR �� random priming �i��Щw�����U�������u�I�H |
||||||||||||||||||||||||||||
| �@ | �@ |
�@ |
| �@ |
�� ���� 3.3 �` �� |
�� TOP ��
�� �̪�q����J 2004/03/23