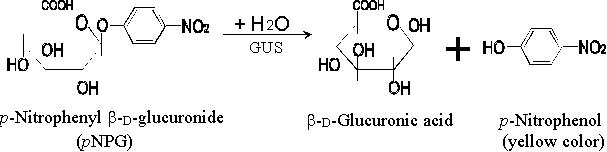Contents �D Chapter 0 �D Chapter 1 �D Chapter 2 �D Chapter 3 �D Chapter 4 �D Appendix
�x�W�j�ǹA�ƨt �� �R ��
| �@ | �@ | �@ |
|
�ͪ��N��k ���@ |
�� �ͪ��N�֤߹��� BCT |
|
|
|
�@�^�ؿ� |
|
| �@ | �@ | �@ |
|
|
�Ĥ@�� | �@ |
| �@ |
Contents �D Chapter 0 �D Chapter 1 �D Chapter 2 �D Chapter 3 �D Chapter 4 �D Appendix |
�@ |
| �@ | �@ | �@ |
| �@ | �ާ@�N | |
| �@ | Basic Laboratory Techniques | �@ |
| �@ |
�x�W�j�ǹA�ƨt �� �R �� |
�@ |
| �@ | �@ | �@ |
| �@ | �@ | �@ | |
| �@ | �@ | ||
| �@ | �@ | �@ | �@ |
| �@ |
1.0 ����Ǧw�� |
�@ |
�@ |
| �@ |
1.1 �L�q�l�ޤ��ϥ� |
�@ | |
| �@ |
1.2 �������p���ϥ� |
�@ | |
| �@ |
1.3 �ӭM���i��k |
�@ | |
| �@ |
1.4 DNA �������Q���R |
�@ | |
| �@ |
�@ |
�@ | |
| �@ |
|
�@ | |
| �@ | �@ | ||
| �@ |
�����ۭ���i�楻�ҵ{�Τ@���������ɡA�ҥ��ݨ�ƪ����ѤΰN���DzߡF�U���綵�ض������㦳�����ʡA���䤺�e�U�ۿW�ߡA�]�A�L�q�l�ިϥΡB���G���t�s�B�������p���ϥΡB���L�ͪ��ާ@��k�A�H�ΰ� DNA ���R��k���C |
�@ | |
| �@ | �@ | �@ | |
| �@ | �@ | �@ | |
|
|
||
|
����Ǧw�� |
||
| �@ | �@ | �@ |
| �@ |
�y�w���z�O�i��������̭��n���Ҷq�A�Y���d�N�A�g�`�|�y���ä[����ѡA�]���A�п��u�H�U�U���W�w�G |
�@ |
| �@ | �@ | �@ |
|
1. |
�Шƥ��ɬd���R�~���B�~���x�η���������m�C |
�@ |
|
2. |
���קK��۲D�c�Ω�c
(�}�k���n�r�S)�A�ýЬ�۹����C�d���v�̡A�b����i��e�̦n�N�Y�v���n�C |
�@ |
|
3. |
�b����ǽФŧl�ҡB���ۡB�Z�f���}�B�Y�F��B�ܶ��ơC�������i�s������Ǫ��B�c���C |
�@ |
|
4. |
����e��бN�u�@�ϰ�M�z�������b�A���礤��½�����ī~�վ��ɡA�n�H�ɲM�z�F���}����ǫe�аO�o�~��C |
�@ |
|
5. |
�ϥΨ�קl�ޮɡA���ťμL�l���A�ХΦw���l�y�Χl�ޮA���C |
�@ |
|
6. |
���ת���P�_�A�̦n��۳Ƥ@�Ʀw������A�t�s���P���G�B���r�����G�ζi��M�I����ɡA�����W����C����Ǹ̦����Ϊ��w������Ψ��@���n�A�Х����x��m����m�C������P�ǡA���קK�����β���ާ@����C |
�@ |
|
7. |
�ϥΥ����ī~�A�Х��ݲM�Хܩ�½�\ Merck index�A�d���O�_�|��H��y���ˮ`�C |
�@ |
| �@ |
a. ���o�ʪ������G �ȥ��b�q���o���q���B�t�s�C |
�@ |
| �@ |
b. ���r�B�P���ľ��G �Ҧp acrylamide (���g�r)�Aethidium bromide (���ܾ�) ������M���ΡA�ýФŨ�B�ìV�C |
�@ |
| �@ |
c. �ī~�o�G�����i�������H�N�ɭˡA�ݨ̫��w�����B�z�C |
�@ |
|
8. |
�ϥλ����ξ���G |
�@ |
| �@ |
a. ���߾��G�۹��m�����ߺްȥ����šC |
�@ |
| �@ |
b. �ѹq���G�|���ͷ������q���A�}�ҹq���ɽФ�IJ�N�q���C |
�@ |
| �@ |
c. �L�i�l�G�[���ɥ�����~�\�κ\�P�}�A�H�K�o���z���C |
�@ |
| �@ |
d. UV �O���ϥΡG�ϥ� UV transilluminator �[����ɡA�ȥ��ϥ� UV �������z�����@�O�β���C |
�@ |
| �@ |
e. �s��O���ϥΡG�Фp�����q�B�ȱi�B��S�H�Φ������������U���C |
�@ |
|
9. |
�߲G���B�z�G |
�@ |
| �@ |
������ϥΤ��ӵ����M���O�f��ߡA���b�Y�DZ��p�U�A����ӵ߬ҥi��y���P�V�C���~�A���礤�����ߺرa���Ӧ۽��骺���ĩʡA�H�N���e���y�����Ҥ����ĩʲӵߪ��c�l�A�G�Ъ`�N�G |
�@ |
| �@ |
a. �P�ӵ߱�IJ�L������e���ΰ��i��A�ϥΫᥲ���g�L���ߤ~�i���C |
�@ |
| �@ |
b. ����Y���p�߳Q�߲G�q��A�ХΤj�q�M���R�~�A�åH 70% �s����r�C |
�@ |
| �@ |
c. �ୱ�Φa�����߲G½�ЮɡA�ХH 10% �}�դ������M�z�C |
�@ |
|
10. |
���רk�P�ǩΤk�P�ǡA�Ҥ��i��W�d�b����Ǥ��C�ίv�����B�믫���n�ɡA�ХߧY�������C |
�@ |
| �@ | �@ |
�@ |
�@�� TOP ��
|
|
||
|
�L�q�l�ޤ��ϥ� |
||
| �@ | �@ | �@ |
| �@ |
�L�q�l�� (micropipet) �O�i��ͤƹ���Τ��l�ͪ��ǹ��窺���Ƥu��A�M�ӨϥΤ�k�����T�P�_�A�H�ηL�q�l�ު��ǽT�ʡA�������v�T�F���絲�G�����T�ʡA�G�й�A�������L�q�l�ާ@�`�J���{�ѡC ������ǩҨϥΪ��L�q�l�ެ� Gilson Pipetman P �t�C�A�]�A P1000, P200, P20 ���C�H�U�ϥΤ�k�Ϊ`�N�ƶ��A���������ۭ�t�Ҫ������ѡA�и�Ū����~�i�����C |
�@ |
| �@ | �@ |
�@ |
|
�{�� Gilson Pipetman P - �L�q�l�ޤ���ѻP�ո� |
�@ | |
| �@ | �@ | �@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
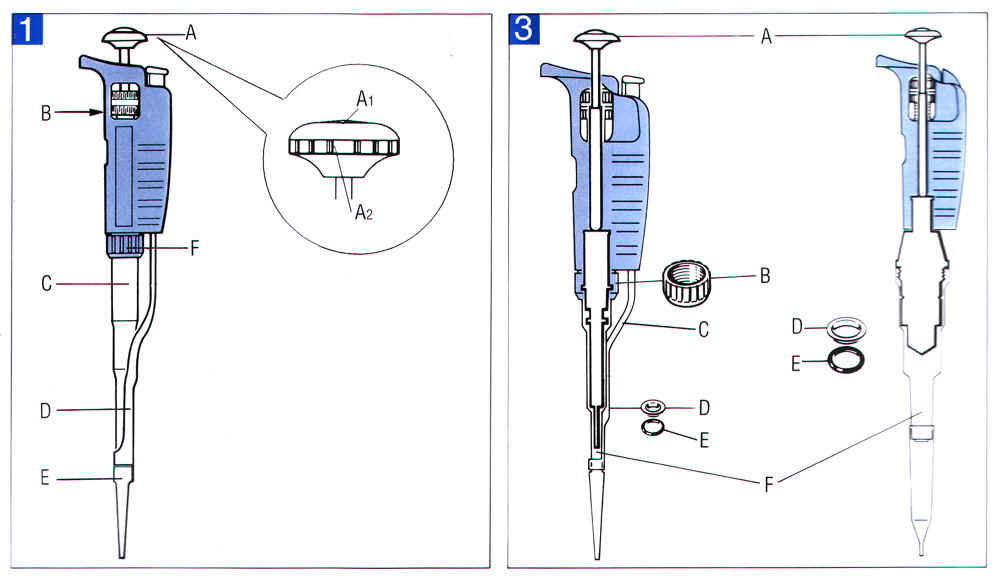
1) �аѷӹ� 1.1�A�{�ѷL�q�l�ިC�@�����ե�m�ΦW�١C |
�@ |
| �@ |
2) �N D (ejector) ���U�AC (connecting nut) �۶}�F�۶}�ɭn�S�O�p�ߡA�] E (piston assembly) �����u®�ܮe���u�X�C |
�@ |
| �@ |
3) �̧DZN C�BH (tip holder) �� E �������U�C |
�@ |
| �@ |
4) �ˬd tip holder ���O�_���ݯd�Q���θվ��C �Y�������A�h�H���M�~���b��A�����B�����C |
�@ |
| �@ |
5) �ˬd E (piston assembly) �W���¦� O-ring (F) �Υզ⪺�K�t�s�� (G, piston seal) �O�_�٦b�H �Y���Q���θվ��ݯd�A�h�H���M�~���b��A�H�s�������A�����C |
�@ |
| �@ |
�� �`�N piston assembly ���i�H�Z�h�L�ίu�Ūo�I��G�C |
�@ |
| �@ |
6) �̧DZN�U�����ե�զX�^�_�쪬�C |
�@ |
| �@ | �@ | �@ |
| �@ | �@ | |
| �@ | �@ | �@ |
| �@ |
|
�@ |
|
�@ |
A: Push-button B: Operating rod C: Connecting nut D: Ejector |
�@ |
|
�@ |
E: Piston assembly F: O-ring G: Piston seal H: Tip-holder |
�@ |
| �@ |
�� 1.1 �L�q�l�� Pipetman P ���ե� (�`���� Gilson Pipetman �ϥΤ�U) |
|
| �@ | �@ |
�@ |
|
�ϥΤ�k |
�@ | ||
| �@ | �@ | �@ | |
| �@ |
1) ��ܾA���� Pipetman�G ���P�������L�q�l�ޡA�U����l����n�d��A�Ш̨��η��G��n���ξA�����L�q�l�ޡC |
�@ | |
| �@ | �@ | ||
| �@ | �@ | �@ | |
| �@ |
2) �]�w��n�G �]�w�^�n�ɡA�ѧC����ܰ��ȡA�����W�V�ұ��]�w�Ȧܤ֤T�����@���A�A����ܳ]�w�ȡF�Ѱ�����ܧC�ȡA�h������ܳ]�w�ȧY�i�C�ФűN�^�n�վ�����W�L�̧C�γ̰����ϥνd��C |
�@ | |
| �@ |
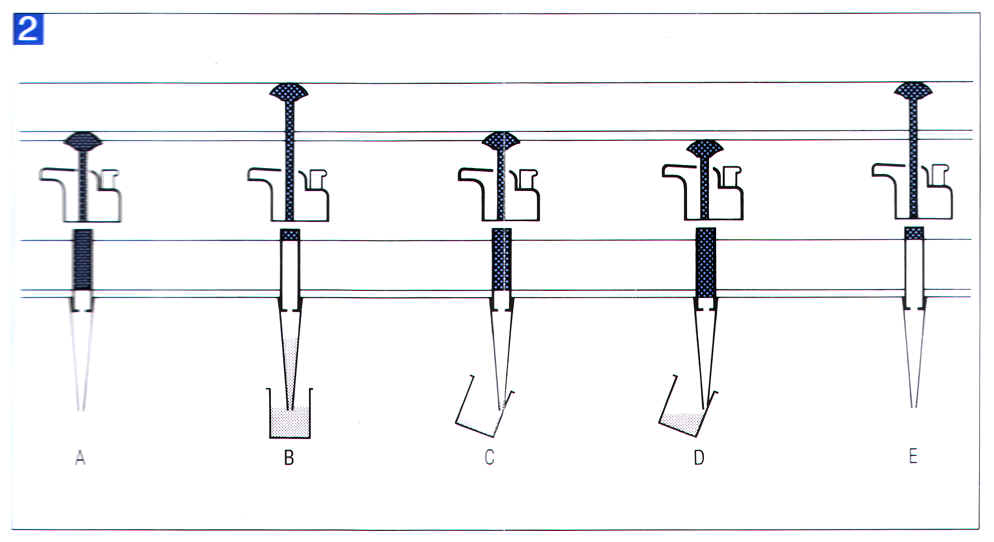
3) �M�W�L�q�l���Y�A�l�����G�G �l�����G�ɡA�y�ݽХ��M�W�L�q�l���Y (tip)�AP1000 �ϥ��Ŧ�L�q�l���Y�AP200 �� P20 �ϥζ���L�q�l���Y�C�N���s���ܲĤ@�q�A���i��O���L�q�l�ޫ����A�N�L�q�l���Y�y�ݮ��J���G�A�A�w�C������s (�� 1.2)�C������s���i�ӧ֡A�H�K���G�ĤJ�l�ެW���ӻG�k����C�L�q�l���Y�y�ݮ��J���G���{���H�l������n�ΨϥΫ����өw�G |
�@ | |
|
�@ |
P10 |
< 1 mm |
�@ |
|
�@ |
P20, P100 |
2 ~ 3 mm |
�@ |
|
�@ |
P200, P1000 |
2 ~ 4 mm |
�@ |
|
�@ |
P5000 |
3 ~ 6 mm |
�@ |
| �@ |
4) ���G�G �N�L�q�l���Y�P�e������IJ�A�C�C���U���s�ܲĤ@�q�A���@����A���ܲĤG�q�A�ⷻ�G�������X�C |
�@ | |
| �@ | �@ | �@ | |
| �@ | �@ | |
| �@ | �@ | |
| �@ |
|
�@ |
| �@ |
�� 1.2 �L�q�l�� Pipetman P ���ϥ� |
�@ |
| �@ |
A, �O���L�q�l�ޫ����A�N���s���ܲĤ@�q�FB, �L�q�l���Y�y�ݮ��J���G�A�w�C������s�FC, �O���L�q�l�ޫ����A�N�L�q�l���Y�P�e������IJ�A�C�C���U���s�ܲĤ@�q�FD, ���ܲĤG�q�ⷻ�G��������X�FE, ������s�^�쪬�C (�`���� Gilson Pipetman �ϥΤ�U) |
|
| �@ | �@ | �@ |
| �@ | ||
| �@ | �@ | �@ |
| �@ |
1) �űN�L�q�l�ޥ�����J���G���C |
�@ |
| �@ |
2) �l���H�װ����ղG�A�Х��N�L�q�l���Y�y�ݥH�M���ΰŤM�N�X�f���j�A�å���w����A�l���C |
�@ |
| �@ |
3) �l���IJG�Ψ�G�k�ʷ��G��A�бN�L�q�l�ީ�Ѷ}�A�U����s��H�]�H���R�~���b�A������A���T�զX�^�_�쪬�C |
�@ |
| �@ |
4) �L�q�l�ު��������ťΤ��N�N�A�礣�i�l���ūװ��� 70�J �����G�A�קK�]��I�J�G�k����C |
�@ |
| �@ |
5) �M���L�q�l���Y���L�q�l�ޡA�L�L�q�l���Y���O�_�����G�A�����i����A�ݪ��߬[�n�C |
�@ |
| �@ |
6) P5000 �����ϥιL�o�Y�A�L�o�Y��çY�ݧC |
�@ |
| �@ |
7) �Y���p�ߨϷ��G�i�J�l�ެW���ɡA�����H��ѡA�N����ե�B�l�ެW�BO-ring�B�K�t�s�Ե��U����H�M���R�~���b��A�A�H�s�������A������A���T�զX�^�_�쪬�C |
�@ |
| �@ |
8) �w���ۦ�H�ѥ��ˬd�ǽT�סA�Y��������D�аe�t���סC |
|
| �@ | �@ |
�@ |
|
�m�ߨϥηL�q�l�� Pipetman P |
�@ | ||||||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | |||||||||||||||||||||||||||||||||||
| �@ |
������G |
�@ | |||||||||||||||||||||||||||||||||||
|
�@ |
Pipetman P1000, P200, P20 |
�@ |
|||||||||||||||||||||||||||||||||||
|
�@ |
�L�q���߾� |
�@ |
|||||||||||||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | |||||||||||||||||||||||||||||||||||
|
�@ |
Solution 1 |
�@ |
|||||||||||||||||||||||||||||||||||
|
�@ |
Solution 2 |
�@ |
|||||||||||||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
1) �Ш�����L�q���ߺޡA���O�ФW A~F�C |
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
2) �H���T�ϥΤ�k�A�̤U���ҦC��n�l�����P���G�ܦU�ޤ��C |
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
|
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
3) �N�\�\�n�A�m�J�L�q���߾��A���Ƭ��Ϸ��G�V�X�ö�����ީ��C |
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
�� ���ߺЪ`�N�n�����Ŧn�I |
�@ | |||||||||||||||||||||||||||||||||||
| �@ |
4) �N�A���L�q�l�ޤ���n�վ��զ��`��n�B�A�H���b���L�q�l���Y�l���ޤ������G�A���ˬd�G |
�@ | |||||||||||||||||||||||||||||||||||
|
�@ |
(i) �L�q�l���Y�y�ݬO�_��n�R�����G�ίd���@�ǪŶ��H |
�@ |
|||||||||||||||||||||||||||||||||||
|
�@ |
(ii) ���ߺޤ��O�_�����G�ݯd�H |
�@ |
|||||||||||||||||||||||||||||||||||
| �@ |
�� �Y�L�q�l�ޥ\�ॿ�`�åB�ާ@���T�A���ߺޤ����L�G��ݯd�A�B�L�q�l���Y�y�ݭ�n�R�����G�C |
||||||||||||||||||||||||||||||||||||
| �@ | �@ |
�@ |
|
�˴��L�q�l�ު��ǽT�� |
�@ | |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
|
�@ |
Pipetman P1000, P200, P20 |
�@ |
|
�@ |
�ѥ� |
�@ |
|
�@ |
�p�N�M�λ]�H�� |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �N P1000, P200, P20 ����n�վ�����̰ܳ��� (�Y 1000, 200, 20)�C |
�@ |
| �@ |
2) �ѥ��W�w����m���įȩί��ĽL���k�s�C�H�L�q�l�ާl���]�H���A�[�b���įȩί��ĽL�W���䭫�q�C�C��L�q�l�ޭ��Ƥ����A�ðO���C�@���o�쪺���G�C |
�@ |
| �@ |
3) �ѵ��G�P�_�A���L�q�l�ެO�_�Ӯե����פF�H |
|
| �@ | �@ |
|
�@�� TOP ��
|
|
||
|
�������p���ϥ� |
||
|
Hitachi U-1100 Spectrophotometer �ϥΤ�k |
�@ | |
| �@ | �@ | �@ |
| �@ |
Hitachi U-1100 Spectrophotometer �ݩ�²�櫬�������A��� D2 �� WI ��ؿO�w�A�i���w���i���d�� 200~1100 nm�A�䵲�c�B��z�Υγ~�и�Ū��t�� Instruction Mannual�C �H�U�ҦC�h����������w���G�l���ת��ާ@��k�A�H�ΨϥΪ`�N�ƶ��C |
�@ |
| �@ | �@ | �@ |
|
�l���״��w��k |
�@ | |
| �@ |
1) ���}�k�e�褧�q���}���C |
�@ |
| �@ |
2) ���}���᰼�� UV Lamp �� VIS Lamp �}���C |
�@ |
| �@ |
3) ��J�i���G |
�@ |
| �@ |
���uWAVE-LENGTH�v�� |
�@ |
| �@ |
���ƭ���A�Ҧp�G�u2�v�u8�v�u0�v |
�@ |
| �@ |
���uENTER�v�� |
�@ |
| �@ |
4) ��� MODE�G |
�@ |
| �@ |
���uMODE�v�� |
�@ |
| �@ |
���uABS�v�� |
�@ |
| �@ |
5) ��J Blank�A���u100% T 0 ABS�v��A�k�s�C |
�@ |
| �@ |
6) ��J�˫~�AŪ���l���ȡC |
|
| �@ | �@ | �@ |
|
�ϥΪ`�N�ƶ� |
�@ | |
| �@ |
1) ���w UV �d��ХΥۭ^�����ޡC |
�@ |
| �@ |
2) ���w���l���ȭY�W�L 2�A�бN���G�}����A���C |
�@ |
| �@ |
3) ���p�W�Фũ�m���~�A�ר�O���G�C��ФŦb���p�W�ާ@����C |
�@ |
| �@ |
4) ��J�����ޫe�A�бN�����ޥ~���������b�A���i������G��ݯd�C |
�@ |
| �@ |
5) �ϥΫ�����ޥ����ߧY�H�]�H���R�~���b�A������m�^���C |
�@ |
| �@ |
6) �ϥΫ�бN���p�����B�����ΩP��M�z���b�C |
�@ |
| �@ |
7) �⦸�ϥζ��j�ɶ��Y���W�L 1 h�A�h�к����}�����A�A���������O�w�ιq���C |
�@ |
| �@ |
8) �ϥΧ����ȥ������O�w�ιq���}���C |
|
| �@ | �@ | �@ |
|
���w���G���l���� |
�@ | |
| �@ | �@ | �@ |
| �@ |
�b�ĥ|�����J�ս�¤ƹ��礤�A�N�|���w b-glucuronidase (GUS) �����ʡA�O�Q�� p-nitrophenyl b-D-glucuronide �����A�[�J�ï�����������X�� p-nitrophenol �b�P�ʤU�e�{���� (�� 1.3)�A�i���w 415 nm ���l���סA�Q�� Beer's Law �Y�i�p��D�o���������ͦ��q�C ������N�Q�Τw���@�פ� p-nitrophenol�A���w�� A415�A�H�D�� p-nitrophenol �b GUS ���ʴ��w����U�� �����Y�� (extinction coefficient)�C |
�@ |
| �@ | �@ | �@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
p-Nitrophenol |
�@ |
|
�@ |
10��GUS assay buffer (0.5 M Na phosphate, pH 7.0; 1% Triton X-100; 0.1 M b-mercaptoethanol) |
�@ |
|
�@ |
GUS �פ�G (2.5 M 2-amino 2-methyl propanediol, �������, pH 11) |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �t�s 5 mM p-nitrophenol stock solution�G ���� _____ mg p-nitrophenol�A�m�� 400 mL �N�M���A�[�J 25 mL 10��GUS assay buffer�A�[���ܬ� 200 mL�A�ͩշ��ѫ�A�[���� 250 mL �òV�X���áC�аt�s�T���C |
�@ |
| �@ |
2) �t�s 50 mM p-nitrophenol ���G�G���O�ѤT�� stock solution ���A�q�H 1��GUS assay buffer �}�����C |
�@ |
| �@ |
3) �� 1 mL 50 mM p-nitrophenol���G��պޤ��A�[�J 0.4 mL 2.5 M 2-amino 2-methyl propanediol ���G�A�V�X���ë���w A415�C �t�� 1 mL 1��GUS assay buffer�A�[�J 0.4 mL 2.5 M 2-amino 2-methyl propanediol ���G�A�V�X���áA����w A415�A�@�� blank�C |
�@ |
| �@ |
4) �� Beer's Law �D�X p-nitrophenol �b�����w����U�������Y�ơC |
�@ |
| �@ | �@ |
�@ |
| �@ | �@ | |
| �@ | �@ | �@ |
| �@ |
|
�@ |
| �@ | �@ | �@ |
| �@ |
�� 1.3 b-Glucuronidase (GUS) �ʤƤ����� |
|
| �@ | �@ | �@ |
�� TOP ��
|
|
||
|
�ӵ߰��i��k |
||
|
��@�߸� (single colony) ������ |
�@ | |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
|
�@ |
�s��O |
�@ |
|
�@ |
������ |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
E. coli K-12 �߮� (JM109) |
�@ |
| �@ |
LB agar plates�G |
�@ |
|
�@ |
1% (w/v) Bacto tryptone, 0.5% Bacto yeast extract, 1% NaCl�A�H 5 N NaOH �զ� pH 7.0 ��A�[�J 1.5% (w/v) Bacto agar�A�H�ü��k���ߡC |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �C�ը��@�� LB plate�A�b���i�ש����Щ��էO�Τ���A�üg�W JM109�C |
�@ |
| �@ |
2) �N�������b�s��O���V�W�N���A�������W�賡����o�H���V�N�L�C�t�@��N�ͪ��� JM109 �����i�\�l���_�ƦT�A�N�������b���i��ťճB���W�Ʀ��H�ϧN�o�C |
�@ |
| �@ |
�� �`�N�I �\�l�Фť��}�θm������W�I |
�@ |
| �@ |
�� �������Фũ�m�b��W�α�IJ��䥦�F��I |
�@ |
| �@ |
3) �H�������������U�߸��A�ñN���i�\�l�m�^�C |
�@ |
|
�@ |
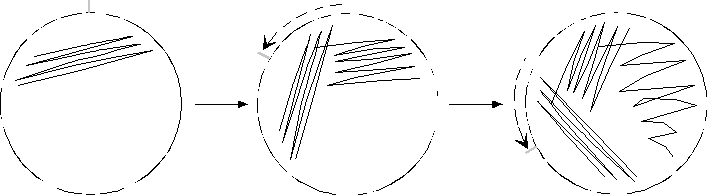
4) ���}�ťժ� LB plate�A�N�g���߸������������W�����s�e�Ʊ���u (�p�� 1.4 �� Streak 1) ��A�N���i�\�l�m�^�C |
�@ |
|
�@ |
5) �A���N�������b�s��O���V�W�N�� �èϧN�o�C �N���i�ױ���� 60 �סA�e�ĤG���u (�p�� 1.4 Streak 2)�C |
�@ |
| �@ |
6) ���ƤW�@�B�J���ާ@�A�e�ĤT���u (�p�� 1.4 Streak 3)�C |
�@ |
| �@ |
7) �N�������b���V�W�N�����߫��i�m�^�����W�C |
�@ |
| �@ |
8) �N���i�˸m�A�� 37�J ���i�c�����i�L�]�C |
�@ |
| �@ | �@ |
|
| �@ | �@ | |
| �@ | �@ | �@ |
| �@ |
|
�@ |
| �@ | �@ | �@ |
| �@ |
Streak 1 Streak 2 Strreak 3 |
�@ |
| �@ | �@ | �@ |
| �@ |
�� 1.4 �������i��@�߸�����k |
|
| �@ | �@ | �@ |
|
�L�]�߲G (overnight suspension culture) �����i |
�@ | |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
|
�@ |
�s��O |
�@ |
|
�@ |
������ |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
E. coli K-12 �߮� (JM109) |
�@ |
| �@ |
LB ���i�G�G |
�@ |
| �@ |
1% (w/v) Bacto tryptone, 0.5% Bacto yeast extract, 1% NaCl�A�H 5 N NaOH �զ� pH 7.0 ��A�H�ü��k���ߡC |
�@ |
|
�@ |
LB agar plates�GLB ���i�G���K�[ 1.5% (w/v) Bacto agar�C |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �C�������@�� 15 mL �պޡA�˦� 2 mL LB ���i�G�A�мg�W JM109�A�üЩ��էO�Τ���C |
�@ |
| �@ |
2) �N�������b�s��O���V�W�N���A���W�賡����H���V�N�L�C�P 1.3.1 ��k�B�J 2) ���ާ@�A�ϱ������N�o�è��U�߸��A�N���i�\�l�m�^�C |
�@ |
| �@ |
3) ���}�պ\ (�@�ⴤ���պޡA�åH���۱����������⪺�p���۶}�\�l)�A�N�ޤf�b�s��O�W�L���C�N�g���߸������������J���i�G���û��V���ϵ߸������C�N�ޤf�A���b�s��O�W�L����A�N�\�l�m�^�C |
�@ |
|
�@ |
4) �N�������b���V�W�N�����߫�m�^�����W�C |
�@ |
|
�@ |
5) �N�պު��\�l�y�L�۶}�A�åH���a�T�w�\�A�H�K�_�����i�ɻ\�l�P�ʱ����C |
�@ |
| �@ |
6) �N�պm�� 37�J ���i�c�Τ��D���_�����i�L�]�C |
�@ |
| �@ |
7) �N���� JM109 �����i�ץ�^���U�СC |
�@ |
| �@ |
�� �P�ӵ߱�IJ�L������e���ΰ��i��A�ϥΫᶷ�g�L���ߤ~�i���C ���礤�Y���p�߳Q�߲G�q��A���W�Τj�q�M���R�~�A�åH 70% �s����r�C�ୱ�Φa�����߲G½�ЮɡA���H 10% �}�դ������M�z�C |
�@ |
| �@ | �@ |
|
|
E. coli JM109 ���ͪ����u |
�@ | |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
|
�@ |
�s��O |
�@ |
|
�@ |
�������p |
�@ |
|
�@ |
�߲G��ٴ� (spreader) �α����� |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
E. coli �߮� (JM109) |
�@ |
| �@ |
LB ���i�G |
�@ |
|
�@ |
LB agar plates |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �C�@�������@�~�˦� 20 mL LB ���i�G���T���@�~�A�мЩ��էO�C |
�@ |
| �@ |
�� �H�U�Ъ`�N�L�߾ާ@�I |
�@ |
| �@ |
2) ���Q�ѱ��بøg�L�]���i�� JM109 �߲G 0.2 mL�A�[�J�T���@�~���A�V�X���ë�A�l�X 1 mL ��L�q���ߺޤ��A�m��B�D���C �T���@�~�h�m�� 37�J �_�����i�C |
�@ |
| �@ |
3) ���C�j�� 30 min �l�X 1 mL �߲G�A���ܤU�ȥ|�I�A�l�X���߲G�Ҽȸm��B�D���C |
�@ |
|
�@ |
4) �C�ը䤤�@�H�бN�� 0, 1, 2, 3, 4, 5 �p�ɤ��߲G�U���X 100 mL �b�L�q���ߺޤ��A�@�t�C�}���F �t�@��P�ǫh�����P�ɶ��l�X�߲G�� A600�A�ХH LB ���i�G�@�� Blank�C |
�@ |
|
�@ |
5) �߲G���t�C�}���G100 mL �߲G�[�J 900 mL �� LB ���i�G�A�V�X���áA���Y�� 101 �}���F�ȩ̀ǧ@ 102, 104, 106 �}���C |
�@ |
| �@ |
6) �N�ťժ� LB plate ���O�Щ��էO�B����B���ˮɶ��ε}���� (�Ҧp�G0 hr, 102)�C �W�z 102, 104, 106 �t�C�}�����߲G�A�U�� 100 mL �[�� LB plate �����C |
�@ |
| �@ |
7) �N�߲G��ٴΥѸ˦��s�몺�N�M�����X�A�b�s��O�W�L���ϴݯd�s�봧�o�A�N�߲G��ٴθm�� LB plate ��t�ťճB�� 30 s �ϧN�o��A�N�߲G��G���áC�R�m�Ƥ����ϵ߲G�����Q���i��l���C |
�@ |
| �@ |
8) �N���i�˸m�A�� 37�J ���i�c�����i�L�]�C |
�@ |
| �@ |
9) �C�@�ɶ��I��߸��ƾA�������i�p��߸��ơA���H�}�����ơA�p���G���C mL ���ơC |
�@ |
| �@ |
10) �H�ɶ��� X-�b�AA600 �εƨ���ƭȬ� Y-�b�@�ϡC �t�H A600 �� X-�b�A�Ƭ� Y-�b�@�ϡC |
|
| �@ | �@ |
|
�@�� TOP ��
|
|
||
|
DNA �������Q���R |
||
| �@ | �@ | �@ |
| �@ |
������O�H���� pBluescript II SK(-) �����ơA�H���P�����Q�@�ΡA�g�q�a���� DNA ���q��A����U DNA ���q���a�ʲv�P���l�q�A�Ǧ��m�߭����Q���ϥΡB�q�a���R��²�� DNA ������� (restriction map) ���إߡC�H DNA �ާ@���D���U�����祲���������G�P�e�������ֻĤ��ѻï��ìV�A�]���ЦP�ǿ��u�H�U��h�þi���ߺD�G |
�@ |
| �@ |
a. ���H�ɫO������ध�M��C |
�@ |
| �@ |
b. �L�q�l���Y�ηL�q���ߺި��X��ߧY�N�\�l�\�W�C |
�@ |
| �@ |
c. �ФťΤ�IJ�N���L�ߪ��L�q�l���Y�B�L�q���ߺޤΦU�خe���������C |
�@ |
|
�@ |
d. ���G���}��A�\�l�Ф��H�N�m��C�l���U�ط��G�Шϥη��L�ߪ��L�q�l���Y�FPipetman �n�O�������A���n�I��e�����F�C���l�X���G��A�ХߧY�N�\�l�\�W�C |
�@ |
| �@ | �@ |
�@ |
|
DNA �������Q���� |
�@ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
������G |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
�@ |
������ |
�@ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�ī~�վ��G |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
���� pBluescript II SK (-) |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�����Q�GBamHI, PvuII �� ScaI (�@�ק��� 10 units/mL, BRL) |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
10��Reaction buffer 6 (0.5 M Tris-HCl, pH 7.4; 60 mM MgCl2; 0.5 M NaCl; 0.5 M KCl) |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�L�ߤ� |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
��k�B�J�G |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
1) �� 7 ��L�q���ߺޡA�̤U���ҦC��n (��� mL) �̧ǥ[������P��m�W�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�� �C���l�������Q�ɡA�д��@��s���L�q�l���Y�H�K�y���ìV�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
|
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�`��n�� 20 mL�A�u�����߫�A�H�L�q�l���Y�����V�X�A#1 �m�B�D���A��l�U�m�� 37�J ���� 1 h�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
2) �m 65�J ���D 15 min ���������A���m�B�D�N�o�A�Y�i�i��q�a���R�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
���ѰQ�סG |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
a. �����Q�G�i�ʦ� Roche Molecular Biochemicals (�Y Boehringer Mannheim Biochemicals), Life Technologies (Gibco BRL), New England BioLabs (BioLabs) �Ψ䥦���q�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
�@ |
b. �����Q���J�s�P�����G ���иѭ�P�ᵲ�O����ï����j�ҡA�]�����i�s���L���B�c���C�j���������Q�ʶR�ӮɧY�O�s�b 50% glycerol ���A�s��b -20�J �ɤ��|�ᵲ�C ���ǭ����Q�۷���í�w�A�����s��b -70�J�A���קK�j�q�ʶR�A�]�й�A�ҥΪ��U�ح����Q���ʽ�@�`�J���F�ѡC �ۦB�c���X�����Q�ХߧY��J -20�J �N�Ჰ�ΦB�D���A�u������ (4�J) �N���W�λ\�W���ï����ߤU��A���ΡC�K�[���P�����Q�����L�q�l���Y�A���藍�i�ìV�C�Ф@�����X�ݭn�q�A�m��L�q���ߺޤ��A�A���O�[�J�U�������F���Ϋ�ХߧY��^�B�c�q�l���Y�ηL�q���ߺި��X��ߧY�N�\�l�\�W�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
�@ |
c. Isoschizomers�G ���Ǥ��P�������Q�i�H�@�Φb�ۦP�� DNA �ǦC�W�A�o�ǻï����٬� isoschizomers�C �Ҧp�JSstI �P SacI�FHincII �P HindII�FEspI �P Bpu1102I�C��������Ƨ��i�Ѥu��ѩΦU�t�ӥؿ��d���ťΤ�IJ�N���L�ߪ��L�q�l���Y�B�L�q���ߺޤΦU�خe���������C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
�@ |
d. �����Q�����������G �C�@�t�ӥͲ��������Q�������̾A�Ϊ� 10 ���@�פ����G (10��Rx Buf)�A�������`�N�A�P�@�ح����Q�Y�Ӧۤ��P�t�ӡA�� Rx Buf �Τ����ūץi�ण�P�A�G�ϥΫe�аȥ��ѦҨ仡���ѩμt�ӥؿ��A�t�ӥؿ������|���C�X�U�ح����Q�b���P Rx Buf �����IJv�C�ϥΨ�إH�W�������Q�i����ήɡA�i�̷ӸӪ���ܾA���� Rx Buf�A�ÿ��`�H�U�X�I��h�J |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
1. �Y�U�ح����Q�ҥΪ� Rx Buf �ۦP�A�h�i�P�ɥ[�J�����G���A�����`�N glycerol ���̲@�פ��i�W�L 10%�A���ǻï��� glycerol �@�ת�������Y��A���i�W�L 5%�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
2. �Y�U�ح����Q�ҥΪ� Rx Buf ���P�A���t���Ȧb�Q���@�ɡA�i���[�J���Q�@���C�̡A���������᪽���[�J NaCl, KCl, MgCl2 ���վ��Q�@�סA�A�[�J�ĤG�ػï��C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
3. �Y�U�ح����Q�ҥΪ� Rx Buf �������P�ɡA�h�H���@�ػï��@�Χ�����A���g drop dialysis ���G�ΥH�s��I�� DNA ��A�A�H�t�@�ػï��@�ΡC |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
e. �����Q�ϥζq�P�����ɶ��G �@��쪺�����Q���ʳq�`�w�q���A�b�̾A�����ūP����U�A��� 1 mg ��� DNA �b 1 h �Q���Χ����һݤ��ï��q�C�q�`�i�[�J���h�q���ï��Χ�����ɶ��[���A�����ǻï���í�w�A�����ɶ��[���ä���N�q�C�[�J���ï��q�ݦҼ{�̪o���̲@�סC�H�קK�̪o�@�פӰ��y����ï����ʪ�����Χ��ܨ�@�ΡC |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
f. ��������G�@�뭭���Q�i�H 65�J �[�� 15 min ��������A�����ǻï�����w�w�ʡA�h�i�[�J EDTA (�̲@�� 10 mM) �ΥH phenol �B�z�C |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
g. �����Q�L�k�@���G |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
���˫~ DNA �L�k�Q���ήɡA�ЧV�O�^�Q���ˬd�G |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
1. DNA �O�_�t�����Q�H |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
2. DNA �O�_�t������H |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
3. Phenol �ΰs��O�_�h�����b�H |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
4. �ï��O�_�J�s�����ӥ��h���ʡH |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
5. ��q�� Rx Buf �O�_�[�J�L�h�H |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
6. �����ūפӰ��ΤӧC�H |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
7. ��O�_�L�k�@�� methylated DNA�H |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| �@ | �@ |
|
|
�v�潦��q�a (agarose gel electrophoresis) |
�@ | |
| �@ | �@ | �@ |
| �@ |
������G |
�@ |
| �@ |
�g�A�q�a�Ѥ�ű���� (Mupid II) |
�@ |
| �@ |
UV transilluminator ���@���n |
�@ |
|
�@ |
��߱o�۾� |
�@ |
| �@ |
�ī~�վ��G |
�@ |
| �@ |
�v�潦 (agarose, low EEO) |
�@ |
| �@ |
1��TAE �q�a�w�IJG (40 mM Tris-acetate, 1 mM EDTA, pH 8.0)�G |
�@ |
| �@ |
�� 50��TAE (�C 1 L�t 242 g Tris base, 57.1 mL glacial acetic acid, 100 mL �� 0.5 M EDTA-8.0) �}���ϥΡC |
�@ |
| �@ |
10�� �l�ܬV���G |
�@ |
| �@ |
0.25% bromophenol blue, 0.25% xylene cyanol, 0.1 M EDTA, 50% glycerol�C |
�@ |
| �@ |
Ethidium bromide (EtBr) stock solution�G |
�@ |
| �@ |
500 mg/mL�A�˩�w�~���A�C 50 mL ���鷻�G�[�@�w (�̲@�� 0.5 mg /mL)�C |
�@ |
| �@ |
DNA�зǤ��l�q�G |
�@ |
| �@ | �@ | |
| �@ |
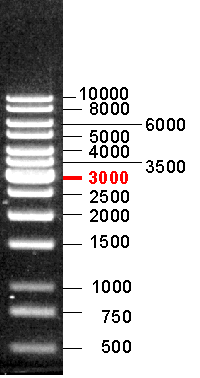
�]�t 8 �Ӥ��q 23.1, 9.4, 6.5, 4.3, 2.3, 2.0, 0.56, 0.125 kb (�� 1.5A)�A�@�� 0.5 mg/mL�A�H�U²�� lMr�C |
�@ |
| �@ | �@ | |
| �@ |
250 bp~10 kb �@ 14 �� DNA ���q (�� 1.5B)�A�@�� 0.5 mg/mL�A²�� LadMr�C |
�@ |
| �@ |
Polarid 667 ���� |
�@ |
| �@ |
��k�B�J�G |
�@ |
| �@ |
1) �C�ջs�Ƥ@�� 12-well 1.2% agarose gel�G �����A�q agarose �����A�[�J 1��TAE (�j�� Mupid II �������� 40 mL)�A�H�L�i�l�[�����ѫ�A�m 55�J ���D���šC |
�@ |
| �@ |
�� Ethidium bromide �����ܾ��A�H�U�ާ@�аȥ�����M�A�øT�����ۤ�M�����B�úN�I |
�@ |
| �@ |
2) �[�J EtBr (�C 50 mL ���鷻�G�[�@�w stock solution)�A�V�X���áA�N���鷻�G�ˤJű���ҡA���W�˥��ޡC�m�Ƿž�����A�p�ߩ}�˥��ޡA�N�����m��q�a�Ѥ��A�ˤJ 1��TAE�A���ܷ��G�\�L�����C |
�@ |
| �@ |
3) �˫~ DNA �� DNA �зǤ��l�q�U�[�J 1/10 ��n�� 10���l�ܬV���C�N�˫~�� lMr �m 65�J ���D 5~10 min�A���ܦB�D���ū�A�Y�i�[�J�������˫~�ѡC |
�@ |
| �@ |
�� LadMr �Фť[���I |
�@ |
| �@ |
4) �H 50V �� 100V �i��q�a�A�ݰl�ܬV�� bromophenol blue ��i�ܽ���T�����G�B�ɡA�����q���A���X�����A�H UV transilluminator box �[���a��m�A�åH��߱o�۾��Ӭ۰O�����G�C |
�@ |
| �@ |
�� �аȥ������@���n�A�H�K���� UV �ˮ`�C |
�@ |
| �@ |
5) �P�зǤ��l�q����A���p�U DNA ���q�����l�q�ëإ߽��� pBluescript ��������СC |
�@ |
| �@ |
�� �t�� EtBr �� agarose gel �Х�ܱM�ήe�����I |
�@ |
| �@ |
���ѰQ�סG |
�@ |
| �@ |
a. ��i�bű���ɤ��[�J EtBr�A�ө�q�a��H�t 0.5 mg/mL EtBr �� 1��TAE �V��A����ؤ�k�ұo�쪺 DNA �a�ʲv�|�y�����P�AEtBr �����[�b�����ɡA�������� DNA ���a�ʲv�|��C�� 15%�C |
�@ |
|
�@ |
b. DNA ���q��ݭY����� (cohesive ends)�A���ɪ��ǦC�|���A�H�X���{�H�C�b�q�a�e DNA �˫~�H 65�J �[����A�A�m��B�D���A�i�Ϩ���H�X���������}������C |
�@ |
| �@ | �@ |
|
| �@ | �@ | |||||||||||||||||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
(A) Lambda DNA/HindIII Marker |
(B) 1 Kb DNA Ladder |
�@ |
�@ | ||||||||||||||||||||||||||||||||||||||||||||
| �@ |
|
|
1
mg
Lambda DNA HindIII Marker �ҧt�U���q���ʤ���ι�ڭ��q
(ng)
|
�@ | ||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�@ |
�@ | ||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
�� 1.5 DNA�зǤ��l�q���զ����q�P�b 1% �v�潦�����q�a���� |
�@ |
||||||||||||||||||||||||||||||||||||||||||||||
| �@ |
(A) Lambda DNA/HindIII marker; (B) 1 Kb DNA Ladder (1 % agarose) |
|
||||||||||||||||||||||||||||||||||||||||||||||
| �@ | �@ | �@ | ||||||||||||||||||||||||||||||||||||||||||||||
�� TOP ��
|
|
||
| �@ |
�ѦҤ��m�G |
�@ |
| �@ |
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith, JA, Struhl K (1987) Current Protocols in Molecular Biology, Vol 1. John Wiley & Sons, Inc, New York |
�@ |
| �@ |
Burton ZF, Kaguni JM (1997) Experiments in Molecular Biology: Biochemical Applications. Academic Press, New York |
�@ |
| �@ |
Hardin C, Presutti D, Miller W, Robertson D (1996) Nucleic Acids, Cloning and Protein Purification: A Laboratory Manual. Simon & Schuster Custom Publishing |
�@ |
| �@ |
Micklos DA, Freyer GA (1990) DNA science: A First Course in Recombinant DNA Technology. Cold Spring Harbor Laboratory Press |
�@ |
| �@ |
Pipetman P, Gilson (operation manual) |
�@ |
| �@ |
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual, ed 2, Vol 1 & 3. Cold Spring Harbor Laboratory Press, New York |
�@ |
| �@ | �@ | �@ |
| �@ |
|
|
| �@ |
Chap1 |
�@ |
�� TOP ��
�� �̪�q����J 2004/03/23