|
2007 進度報告摘要 |
|
| 07/12/12 |
1 林 之 儀 甘藷 L-SP 之 PI 活性催化機制 |
| one-page |
|
| summary |
1. 重新檢討 primer independent 下將 Glc-1-P 置換成 Glc 及 maltose 的實驗,並盡快獲得 L-SP'。 2. 探討 L-SP 是否與 GP 一樣有異位調控機制。使用酵素動力學方法,來觀察 GP 活性之異位調控抑制劑 (caffeine 等) 對 L-SP 之作用。 3. L-SP* 取得困難,需尋找可迅速降解 L-78 片段的方法。 4. 硫酸銨分劃所取範圍可以增加至 20%~60% 以獲得較多 L-SP。 5. 若以全長 L-SP 及部分降解的 L-SP 片段做結晶,是否會造成繞射結果不清楚? 6. L-SP 純化過程應加入適量蛋白酶以抑制降解。
Future work: 1. Purify more L-SP, and add protease inhibitor or glycerol to prevent degradation. 2. Examine the effect on L-SP by replacing Glc-1-P with Gal-1-P (C-4 epimer) or Man-1-P (C-2 epimer). 3. A specific inhibitor to GP might be used to check its effect on L-SP. Because of its flexibility and unstable structure, employ Glc-1-P or its analogs to react with L78, in order to stabilize and maintain the conformation. 4. Amylose purification to explore the effect of plastid. |
| 07/11/28 |
1 羅 庭 芳 比較不同本土禽流感病毒株之蛋白質體與毒性強弱之關係 |
| one-page |
|
| summary |
1. 再研究清楚王老師發表 2838K 及 2838N 的兩篇論文內容,以及 HA 蛋白的歷史。 2. 知道全部 sequence 後應做蛋白質結構比對,特別是 binding site 部份,預測兩株 H6N1 和 sialic acid (SA) 的結合結構 (docking),特別是 HA 是藉由哪一個胺基酸上的醣和 SA 結合。 3. 目前想法是因為 HA 醣基化的不同,改變了對 SA 的結合能力,進而影響到 HA 的感染力及標的器官;但也很有可能是從 2838N 突變成 2838V 後,HA 胺基酸有 1.41% 的改變,直接造成了蛋白質結構的改變,使表面的醣基角度更適合和 SA 結合,而非此兩株蛋白質醣基化之不同。 4. 醣晶片的 sample 要多樣化,可做一組 crude 以及一組由電泳溶離出來的 sample 互相比較。 5. 免疫染色的結果 (與何杰龍結果比較) 差異過大,應再重複幾次實驗。 6. 是否能由王老師取得病理切片,並以抗體進行組織染色。另外亦可使用細胞株進行免疫染色,以螢光顯微鏡或流式細胞儀偵測,看 2838V 的 HA 是否真的對腎臟有攻擊性,並了解腎臟上的 receptor,以推知 HA 和腎臟細胞結合的專一性。
未來工作 1. Refine the protocol for protein precipitation 2. Repeat immunoblot analysis of AIV proteins 3. Bio-mass spectrometry to identify key spots 4. Identification of the AIV posttranslational modification 5. Identification of the glycosylation by glycan microarray technology |
| 07/11/21 |
1 賈 儒 珍 甘藷 L-SP 重組蛋白之表現 |
| one-page |
|
| summary |
1. 再 review 瓊尹 data,確定 SPC 是否具有合成方向的活性。 2. 造成 SP 降解的主要因子可能是 proteasome? (由宏祥的數據推測) 3. 現階段主要工作是拿到 SP 全長,期望能得到有活性的 SP 表現蛋白,之後以此為工具,進行 PI activity 和 SP 降解路徑的研究。 4. 目前得到的 SPN cDNA,所轉譯出的胺基酸序列與已發表序列有 5 處不同,其中有兩處位於 L78 上,無法用於 SP 全長的表現載體構築;得到此段序列的可能是 PCR 造成的錯誤,或是 L-SP 在基因體中不只有一個 copy 數。 5. L78 cDNA 的 GC content 只有 35%,帶有 L78 的序列不太穩定,一直很難構築入載體。可嘗試一次 PCR 出全長再進入載體,或在不影響 codon 前提下把 L78 上幾個 A 或 T 改成 G 或 C (不影響最後的胺基酸),增加 L78 序列穩定度。 6. L78 cDNA 已進入保存載體,但表現的長度要增加到 100 個胺基酸。另外也要構築 SP 全長不含 L100 的表現載體。 7. LSK 對研究 SP 的磷酸化和降解路徑很重要,應試著得到 LSK 的序列並表現蛋白。
未來工作: 1. 持續構築 SP 和 L100 的表現載體,並得到有活性的表現蛋白。 2. 釣取 LSK cDNA。 3. 利用 SP mutant 進行 PI activity 和 SP 降解路徑之研究。 |
| 07/11/14 |
5 林 怡 岑 D-enzyme 的選殖與活性表現 |
| one-page |
|
| summary |
1. 整理 D-enzyme knock-out 相關文獻,並查閱 D-enzyme 與 L-SP 可能共同作用的報告,以推測 L-SP 和 D-enzyme 構成蛋白質複合體後,可能扮演的生理角色。 2. 目前 L-SP 與 D-enzyme 可能形成蛋白質複合體之證據:(a) 免疫共沉澱實驗、(b) Blue native/SDS 2-DE 之次單元組成,以及 (c) 高分子量活性色帶之相似性。 3. 在 E. coli 中 D-enzyme 與 L-SP 位於相同的操縱子 (mal4 operon),受到相同的調控機制,那麼在真核細胞中是否有類似的調控方式? 4. 如何排除 D-enzyme 與 L-SP 之結合,乃因於非試管中聚集所造成的假象? 5. 可利用 D-enzyme 的兔子抗血清進行 double diffusion 試驗。 6. 利用酵素動力學及產物結構測定,觀察外加 D-enzyme 或 L-SP 是否會使 D-enzyme 或 L-SP 的活性變高,及此複合體可能在生理中的作用模式。 7. D-enzyme 之所以可能將短鏈 MOS 變成長鏈 MOS,然後交給 L-SP 作用,是由於 L-SP 進行磷解反應時,所需的最短糖鏈為五糖。 8. 需進一步證明磷酸化或去磷酸化作用,是否對於 HX 有影響,並確認 HX 中的 L-SP (110 kD) 是否受到磷酸化或醣基化修飾。 9. 製備 proteasome 及 D-enzyme 的兔子抗血清,以進行組織定位免疫金標定。 10. 需找出破菌最適條件,使可溶性蛋白質量增加,並具有活性。 11. 比對 D-enzyme 蛋白質活性區位置,並觀察是否具有保守性。 12. 整理維德及宏祥所作的關於磷酸化 L-SP, proteasome 及 inhibitor 相互反應的實驗。宏祥以純化的 proteasome 添加 L-SP (磷酸化),發現磷酸化可能促進 proteasome 對 L-SP 的降解 (slide)。 |
| 07/10/31 |
18 王 信 傑 PCS 的催化機制:磷酸化 |
| 07/11/29 |
1. PCS-N (At221) 有 PCS 活性,此活性可被磷酸脢 CIAP 去磷酸後抑制 50%,磷酸化後可恢復。 2. 鎘加入的先後次序對 At221 的活性有影響,在最後才加入時,才有最正常的活性表現。 3. 全長的 AtPCS 則對 CIAP 較不敏感,只有大約 15% 抑制,但也可被磷酸化所恢復。 4. 用抗 P-Tyr 抗體可以染到表現的 AtPCS,但是抓不到 P-Ser,而磷酸化是用 CKII 做的。 5. 把結果以正式論文圖表方式一一列出,能否說一個完整故事,看還欠什麼關節重點。 |
|
1. 確定表現蛋白 AtPCS 被磷酸化,磷酸化位置位於 N-terminal 靠近 substrate binding site 的地方。目前已預測出可能磷酸化位置,接下來要用 mutagenesis 確認。 2. 直接進行磷酸化對 PCS 活性無大影響,但若先加 Cd 後再磷酸化則會抑制 PCS 活性,推測 PCS 可能需由 Cd 變化構形,才能藉由磷酸化來調控活性。[再仔細檢查此點] 3. 必須再設計其它實驗指出 PCS 磷酸化的生理意義。例如 in vivo 是否有磷酸化?能否純化出磷酸化 PCS?[不容易] 磷酸化 PCS 對細胞之生理影響等。 4. 可嘗試 phosphoprotein purification kit 純化磷酸化 PCS [enrichment],再利用抗體來辨認,不過生物體內 PCS 含量可能非常少,必須要大量純化。 5. 目前為止,純化 endogenous PCS 與定序經驗指出,PCS 在生物體內含量非常少,而且純化不易。雖然量少可是一旦活化後活性卻是非常強,這的確符合經濟效益,這也就是為什麼到目前為止還沒有人可以大量純化出 PCS。所以若能純化出 endogenous PCS 就能針對其生理意義加以探討。當然首要瞭解 PCS 的特性,與有效純化方法。[能否用抗體親和吸著劑通過大量樣本以吸附足夠量的 PCS] 6. PCS 磷酸化後偵測活性產物應該也要測 PC3, PC4 等,不能只看 PC2 而已 [為什麼?]。另外 PCn 結果應標準量化並加入標準差。 7. PCS 磷酸化意義目前有以下幾個思考方向: a) 會不會影響 PCS 催化方向? 合成或分解? [因為基本上 PCS 是蛋白脢] b) 抑制 PCS 功能。[如上條 2] c) 除了合成 PC 外,PCS 還具有其他功能可受到磷酸化調控? 8. 除了 AtPCS 與 At221 外,Nt1 與 Nt2 也要表現出來,對 2nd binding site 能有更多研究。 9. Review 目前已發表之 PCS mechanism 相關文章,讓大家瞭解 PCS 研究進展。 10.根據已發表論文及自己的結果感覺,2nd substrate binding site 的存在越來越可信。目前推測 PCS mechanism 有兩種 models:[下次報告請再說明] a) Acylation reaction:可能在 N-terminal 處的 C138 或 C144 進行,須經由 mutagenesis 來進行研究。 b) Conformational pool:PCS 2nd substrate binding site 是經由構型所形成的 pool,可以讓不同長度 PCn 分子進入。 目前較 favor 第二種 model。若能藉由 Nt1 與 Nt2 表現,再加上 proteinase K 相關研究,針對此問題得到初步解答,最直接證據就是利用放射性 PCn 當基質。 11. PCS 可能具有 substrate specificity。2000 年吳建興博士利用部份純化 PCS 與 PC2 反應可以得到 PC3 與 PC4 產物。2005 年 Hirata 利用表現蛋白 AtPCS 與 PC2 反應也可以得到 PC3 與 PC4 產物,因此認為 PCn 也可以當成 donor molecule。但是本實驗卻發現不論是用 PC2 或 PC3,AtPCS 都無法合成 PCn。關於這點還要再進行確認。[有問題?] 12. 本篇論文可能的創新點: a) PCS 可以被磷酸化,但是磷酸化之生理意義為何? (持續探討中) b) 2nd substrate binding site 的存在與位置。 c) 擁有 PCS 的抗體。[統計目前有幾篇論文製備得抗體?] d) 具備純化 endogenous PCS 的能力 (部份純化)。 e) 若能善加利用以上發現與工具,導入 in vivo 實驗,則本論文會更加完善。 |
|
| 07/10/17 |
8 何 杰 龍 AIV 蛋白質體之探討 |
|
1. 擬測試由諾貝爾提供之 Q proteome GlycoArray,初步檢測 HA 上 N-glycosidic linkage 醣基化的位置。但應先瞭解其基本原理及步驟,解釋為何能分辨不同 glycan ? 2. 將 AIV 2-D gel 以 PAS 或過碘酸-硝酸銀染色法進行初步醣蛋白之分析。 3. 再次以 N-glycosidase 進行 AIV 蛋白質 deglycosylation,另外以 phosphatase 去磷酸。 4. 設計 NA 關鍵 peptides 進行免疫,並持續擴充 AIV 單株抗體庫。 5. 要安排好就職工作與本身論文實驗的時間分配。 6. 與王老師討論,擬將已有 AIV 單株抗體應用於病理切片之組織免疫染色。 |
|
| 07/08/29 |
2 劉 雨 亭 L78P 的立體構造與 L-SP 催化活性關係研究 |
|
1. 請標明 L78 sequence 插入在 SP 模型中的位置。 2. E. coli BL21 前面學名需斜體。 3. 請跑 crude extract 電泳圖。 4. 下次請進行 CD 及 NMR 的原理解說。 5. 因為需大量純化,使用 FPLC 壓力過大,可考慮改用 LC 代替。 6. 可嘗試將多次培養的菌離心後合併在一起,累積至夠量後一次破菌純化。 7. CD 在看結構上有多大的意義?如何在 CD 的結構預測表中計算其百分比? 8. L78 應表示為 L78P。 9. 可進行 L78與 Glc-1-P 親和力測試,比較 Glc-1-P free form 及結合 form 的量。 11. 可詢問楊健志老師有關分子模擬軟體使用的問題。 12. 查詢 TFE 結構穩定劑的可行性與原理。 13. 人工合成 L78 部份 peptide,檢測與 Glc-1-P 結合能力,或對 PI 活性的 rescue。 |
|
| 07/08/15 |
1 眭 毓 庭 建立黃麴毒素的專一性抗體免疫分析方法 |
|
1. 抗體效價的作圖,要將曲線做完全,最好有四重複,並且要標出 deviation。 2. 關於 ELISA 的 control 組,要設計多組控制組,才能確定各個步驟沒有問題,不能只是以 NET 作為補體積用而為 negative control。 a) 不 coatig antigen,但要加一抗及二抗 (negative control)。 b) 不加一抗 (negative control)。 c) Coating 一抗,加二抗 (positive control)。 d) Coating BSA,便能確定是否會對 BSA 有 cross-reaction。 3. 用未經免疫的小鼠血清作為一抗,並逐週測 ELISA 效價,便可看出效價成長變化。 4. AFB1-KLH 跑電泳無法成功的原因,可能是因為: a) Loading 的 sample 量不足,以致染色看不出來。 b) AFB1 難溶,sample 有沉澱,卡在 well 裡下不來。可用超音波震盪懸浮沉澱。 5. 可以試用 dot blotting 取代 western blotting,避免電泳跑不下來的問題。 6. 未來工作: a) 作 competitive ELISA。 b) 繼續解決 AFB1-KLH 電泳的問題。 c) 接續之後的 AFB1 含量測試。 d) 如何做出可分辨 B1 及 M1 的 ELISA? |
|
| 07/07/18 |
2 沈 志 昱 哺乳類胚胎發育過程的關鍵蛋白質 |
|
3 ▲ 1 |
1. 第一階段合成之三種目標蛋白質 (AK, BC, NM),必須清楚個別生理功能,並找出原始文獻。 2. 多多留意各家合成 peptide 公司,選一家有信譽且品質穩定的公司,確保後續實驗不會因為合成之 peptide 序列錯誤,導致實驗產生問題。 並思考如何自行檢測合成 peptide 之正確性? 3. 小鼠、豬與人類 gene homology 程度很高,如欲以小鼠製備抗體,因同是哺乳動物,恐怕有很大的問題。因此應該改以鳥類 (母雞) 作為免疫物種,製備 IgY 抗體,待確定有效價後,直接以小鼠胚胎進行 2-DE 檢測,期望做出能檢定目標蛋白質的抗體。 4. 根據先前 2-DE 結果預估,一次 2-cell 期 2-DE 所需胚胎數,須從 72 個提高至 200 個。以各十片 2-DE 預估,總計約需 2-cell 期胚胎 2000 個,4-cell 期胚胎 1000 個。目前正繼續累積胚胎數量。 5. 再多多搜尋各種可能與繁殖力相關的蛋白質,並儘早完成 peptide 合成。 |
| 07/06/28 |
2 蔡 和 成 以全抗體差異吸附法檢定竹筍生長關鍵蛋白質 |
|
報告摘要: 1. 進行 mouse 抗體庫抗體篩選,初步篩選出來的抗體,專一性及效價皆不甚理想。 2. 另尋其它方法,於是進行兔子免疫,得到對應竹筍全蛋白的 pAb。 3. 針對 rabbit pAb 設計一些相關實驗,其目的都是想要得到具消長之蛋白質相對抗體,用於生理探討。
建議事項: 1. Rabbit pAbs 效價測定 (Western): a) 舊的 anti-rabbit 二抗年代久遠,需購買新的二抗。 b) 二抗稀釋倍率改成 5,000 倍。 c) 加入單獨只有二抗的 negative control。 2. pAb 純化所用的緩衝濃液系統,Tris 濃度可能太低,建議改用 PB 系統。 3. 實驗計劃流程圖會讓人家誤解,需再改過。
未來工作: 1. 利用 rabbit pAb 進行 affinity column 製備,欲利用 affinity column 進行 subtraction,進而得到 BS0 及 BS40 specific protein,以用於抗體庫建立。 2. 利用 BS0 & BS40 2D western 比較,選取差異點進行 peptide 設計,用於老鼠免疫以生產單株抗體。 3. 利用 rabbit pAb 及 mouse mAb 進行 sandwich ELISA,想要直接得到消長蛋白質的相對抗體。 |
|
| 07/06/27 |
X 林 怡 岑 學位資格考試 (答問錄) |
|
Q: 電泳膠片中加入 glycogen,D-enzyme 的泳動率是否有改變? A: 觀察結果似乎沒有改變。 Q: 純化之 HX 內是否含有 H-SP? A: 由 H-SP 活性染色觀察的結果,HX 內應該不含 H-SP。 Q: D-enzyme 的活性染色方法為何?為何染色結果如此模糊? A: 由於 D-enzyme 可以將 maltotriose 轉變成 Glc,接著利用 hexokinase 及 glucose-6P dehydrogenase (G6PDH) 耦合反應,產生 NADPH,使 MTT 與 PMS 轉變為藍紫色沉澱物;可能由於 D-enzyme 含量過低,造成染色結果不明顯。 Q: 在 12.5% SDS-PAGE 及 native/SDS-2DE 中都可以看到 D-enzyme 除了有 60 kDa 的蛋白質色帶外,還有另一個約 180 kDa 之蛋白質色帶,為何在 IEF/SDS-2DE 中只有 60 kDa 的蛋白質色帶,D-enzyme 是否有不同的 form? A: 約 180 kDa 之蛋白質色帶可能是第二維電泳之前處理劇烈程度不同,造成 trimer 或是 monomer 與其他蛋白質結合之結構未被完全破壞所造成。 Q: 可試著以 IEF pH 3-10之 2-DE 觀察是否有不同 form 的 D-enzyme。 Q: 是否有比對過不同物種 D-enzyme 的相似度? A: 比對過阿拉伯芥和馬鈴薯的 D-enzyme,相似度大約 68%。 Q: 免疫沉澱實驗中 LC/MS/MS 的結果除了有定到 D-enzyme 之外,是否還有定到其它的蛋白質? A: 沒有。 Q: 2-DE 中,與 D-enzyme 相同分子量之蛋白質身分為何? A: 經 LC/MS/MS 定序的結果為 chaperonin。 Q: 2-DE 中,L-SP 之分子量為何有明顯的改變? A: HX 中至少含有兩種形式之 L-SP,一種是與 proteasome 及 D-enzyme 結合的形式,另一種是原態的 L-SP,所以分子量的改變可能是由於不同形式的 L-SP,或 L-SP 遭受降解所造成。 Q: 圖 3.13C, D 為同一張轉印膜嗎? A: 是的,先將轉印膜以二次抗體免疫呈色 (圖 3.13D) 後,再以 H7c 進行第二次免疫呈色 (圖 3.13C)。
Q: 目前有 paper 證明 L-SP 和 D-enzyme 會互相結合嗎? A: 沒有,但是有推論認為兩者是連續性的反應 (conjunction)。 Q: 為何推論兩者是連續性的反應 (conjunction)? A: 因為兩者在 E. coli 是由同一個操作組所調控。 Q: 但是 E. coli 與高等植物的醣類代謝路徑應該不同。 Q: 如何定義蛋白質之間是 conjunction 或是 interaction? A: 如果 A 與 B 混合後,A 或 B 的活性有增加,就可以支持兩者為 conjunction,而需要有IP、yeast two-hybrid 及 pull-down 等實驗證據,才能推論兩者互相結合。 Q: 請舉出三個蛋白質與生物分子具有交互作用的例子? A: Antigen-antibody、enzyme-substrate 以及 recepter-ligand。 Q: 以上三個例子何者的親和力較強? A: Antigen-antibody 應該比 enzyme-substrate 強。 Q: 描述抗原抗體間的親和力所用的單位為何?大約多少? A: Kd。大約 10 的負 10~14 次方 (mM)。 Q: 請舉出生化課本中,蛋白質以複合體形式存在的例子? A: Pyruvate dehydrogenase。 Q: 未來打算再進行那些實驗? A: 計畫將 L-SP 及 D-enzyme 表現出來進行 pull-down assay。 Q: Immunoprecipitaton 已經是一種 pull-down assay。 Q: 免疫沉澱實驗中,不同鹽濃度流洗,能決定蛋白質之間的結合是專一性還是非專一性,請問你是用多少鹽濃度的緩衝液流洗? A: 以 PBST 流洗,鹽濃度為 0.15 M NaCl。 Q: 甘藷沒有 cDNA libarary,你要如何表現 D-enzyme? A: 用 RACE,一端用 conserved primer,另一端用 universal primer mix。 Q: 最好能設計兩段 conserved primer 來進行。 Q: 如果蛋白質量夠多可考慮用光譜的方式去證明蛋白質之間互相結合。 Q: 可否利用 TLC 去偵測 D-enzyme 的活性? A: 可以,很多文獻都有利用此方法。 Q: 第二張投影片中所描述的是夜晚阿拉伯芥葉片中的澱粉降解路徑,甘藷塊根中有類似的代謝路徑嗎? A: 應該不太一樣。 Q: 請解釋在不同組織中對於澱粉代謝有哪些不同的生理需求? A: 在葉片中,葉綠體會在白天合成短暫性澱粉,到了夜晚,澱粉粒經由一連串酵素作用而降解,以供細胞生長及能量需求;而在塊根或塊莖組織中所產生的儲藏性澱粉是長期性儲存碳源的場所;然而在種子中的儲藏性澱粉則是作為種子生長發育用之長期性碳源。 Q: 可否根據一些 review,提出適用於甘藷塊根澱粉代謝路徑之假說? A: 目前文獻所推論的兩種假說,一為兩者共同作用修飾澱粉結構,二為重新利用三糖以合成澱粉都有可能。 Q: b-Amylase 對四糖的水解效率已經很低,但是四糖對於 L-SP primer-independent 活性卻迅速增加,這之間有何關係? Q: Amyloplast 和 chloroplast 有何關係? A: 同源胞器。
Q: 請問效價的定義是什麼,論文中所寫的「測試效價」這樣的描述正確嗎? A: 效價的定義為將抗體序列稀釋後,最大吸光值之一半所對應的抗體稀釋濃度即為此抗體之效價,論文中所寫的「測試效價」並不正確。 Q: HX 外加 CIAP 的實驗中,如何證明不是 protease 所造成的結果? A: 此 CIAP 是commercialized,廠商標榜 protease free。 Q: 有沒有其他方法可以證明不是 protease 所造成的結果? A: 看 SDS-PAGE 中,SP110 有沒有斷裂。 Q: 其實可以不要加 CIAP 所需的金屬離子,或是加入 CIAP 的 inhibitor 作為控制組,請問CIAP 所需的金屬離子為何? A: Mg2+ 離子。 Q: 論文中有時候寫 D-enzyme 有時候寫 DPE1,兩者為同一蛋白質,文字上應統一描述。 Q: 在以 D-enzyme 單株抗體分析膠體過濾法之各分劃蛋白質中,為何 D-enzyme 顯示有三種分佈,甘藷中有幾種 DPE? A: 目前推測一種是 D-enzyme 單體的形式,一種是 D-enzyme 和 L-SP 結合的形式,另一種則為 D-enzyme 和 L-SP 複合體經電泳處理而解離所造成;甘藷應該只含兩種 DPE,不確定此單株抗體是否兩種 DPE 都會辨識。
Q: 如果要再做免疫沉澱實驗,你覺得用共價性還是非共價性接合抗體好? A: 共價性接合抗體,因為可以避免抗體色帶的干擾。 Q: 但是非共價性接合抗體的效果較好,所沉澱的蛋白質量較多。 Q: 為何免疫沉澱的結果中,L-SP 和 D-enzyme 沉澱的蛋白質莫爾數比例差不多,可是proteasome 的蛋白質量卻明顯少很多? A: ………………。 Q: 如何以 ELISA 篩選 D-enzyme 抗體? A: 以部分純化之 HX 進行 ELISA,由於抗原複雜所以只能作為初步篩選,需經由 1D Western 作進一步的確認。 Q: 掃瞄時所出現的亮線要如何解決? A: ………………..不知道。 Q: 可能是一開始掃描的光源處有水滴所造成,或者是機器內部進水。 A: 謝謝老師告知。 Q: D-enzyme 經由 LC/MS/MS 定序的分數都很低,原因有兩個,一個是蛋白質量太少,一個是 homology 太低,妳覺得主要的原因是哪一個,理由是什麼? A: Homology 太低,因為蛋白質量少有時分數也很高。 Q: 理論上比對到一條序列,分數就有 60-70 分以上,D-enzyme 比對到兩條序列分數還那麼低,很明顯是因為 homology 太低。 Q: D-enzyme 的活染色帶強弱與蛋白質量有關,要改進初步純化中 D-enzyme 的活染圖譜可能不容易。 Q: Primer-independent 實驗中的 L-SP 會不會含有 D-enzyme,導致 L-SP 合成為三糖時,由於 D-enzyme 幫助,使澱粉含量迅速增加,此假設可經由 primer-independent 實驗外加HX 來證明。 Q: L-SP 和 D-enzyme 互相結合可促進 L-SP 合成澱粉,L-SP 和 proteasome 互相結合可促進 L-SP 磷解澱粉,而三者竟然剛好在同一分劃中出現。 Q: L-SP 和 D-enzyme 之複合體似乎在澱粉代謝上扮演重要的功能,值得深入去探討。 Q: 在 chloroplast 中已經確定有 b-amylase,那在 amyloplast 中沒有嗎? A: 目前的研究仍不確定 amyloplast 中是否含有 b-amylase。 Q: 請作出一張工作完成進度表,定期向口試委員報告。
Q: 妳覺得 HX 到底是誰跟誰互相結合所造成? A: 由於 L-SP 高分子量色帶不只一條,所以 L-SP 與 proteasome、L-SP 與 D-enzyme、或 L-SP、proteasome 與 D-enzyme,這三種結合都很有可能。 Q: 也有可能是 L-SP 和 D-enzyme 與同一條 amylose 結合所造成的結果。 Q: Teltow 是如何證明 L-SP 和 SBE 的結合需要磷酸化作用? A: 先分離純化出 amyloplast,接著在 amyloplast 萃取液中加入帶有放射線 ATP,並進行免疫共沉澱實驗,結果發現免疫共沉澱後的蛋白質都帶有放射線。 Q: 需進一步證明這些蛋白質結合作用在生理上是有意義的,而不是細胞打破後,蛋白質聚集所造成的假象。 Q: D-enzyme 經由 LC/MS/MS 定序的分數都很低,原因為何? A: 可能有兩個原因,一個是蛋白質量太少,一個是 homology 太低。 Q: 如何去確認 2-DE 上的蛋白質位移是何種修飾所造成? A: 一種方法是利用 anti-phospho-Ser/Thr/Tyr 抗體進行免疫染色,另一種方法是將 HX 經CIAP 處理後,比較處理前後 HX 之 2-DE 圖譜。 Q: 這是針對磷酸化修飾,有沒有可能有其他修飾作用? A: 也有可能是醣基化修飾。 Q: 有些 D-enzyme 的活性染色圖譜很模糊,有沒有辦法改進? A: 假若提高 D-enzyme 的蛋白質量,活性色帶會比較明顯。 Q: 為何 D-enzyme 要將短鏈 MOS 變成長鏈 MOS,才能給 L-SP 作用? A: 因為 L-SP 進行磷解反應時,所需的最短糖鏈為五糖。 Q: D-enzyme 只能對三糖作用嗎? A: 三糖以上都可以,但是對三糖的親和力最強。 Q: 假若 L-SP 與 D-enzyme 互相結合,那麼此複合體所扮演的生理功能為何? Q: L-SP 可能扮演控制醣類代謝方向的調控者,根據不同的生理需求,合成或磷解澱粉。 Q: 未來工作所寫的都是實驗方法,沒有靈魂,應該畫一張具體的流程圖,並提出對於甘藷塊根澱粉代謝路徑可能的假說。
|
|
| 07/06/21 |
2 饒 驤 (甘藷澱粉磷解酶 H-SP 之純化與催化機制) |
|
1. H-SP 的純化需更加熟練,才能使產率更好。 2. 每一個純化步驟都一定要進行 Bradford 定量與 CBR 染色,不能省略。 3. 活性分析與蛋白質定量至少每 3 個 fractions 就要測一個數據點,才不會不準。 4. 當樣本通完 ConA (I) 後活性不見,或許 H-SP 的某部分因子 (可能是醣類) 被 ConA 吸走,又或許 H-SP 仍結合於 ConA (I),並未 flow through下來。(Meeting 後老師給我的建言:以後的純化省掉此步驟。) 5. 比較 primer-independent 活性時,實驗設計需更仔細些,需做更明確的各反應物濃度及時間點的記錄。 6. 硫酸銨分劃的活性電泳圖不足以準確判定活性高低,因為每一個分劃中含有各種雜質,有些會抑制活性,有些可能還會與 H-SP 形成複合物,會影響到活性。 7. 我設計的親和性製備式電泳實不可行,原因大致如下: a. 製備式電泳的回收率很低,且 H-SP 本來就很少。 b. 可能電壓會太高,會讓膠片太熱,使蛋白質失去活性。 c. 雖經長時間電泳,但可能還有很多雜蛋白沒電泳出膠片外。 d. Acrylamide 價格不便宜,用此方法純化很花錢。 e. 雖然出現好幾條活性色帶,但因為這是用大片膠所呈現,所以比較不準。 f. 上述出現許多條活性色帶的現象,可能由很多原因造成,短期內很難研究清楚,不建議碩士生來進行這部分的研究。 未來工作: 1. 繼續 H-SP 的純化,以助更瞭解其性質。 2. 再多查一些有關 H-SP 的資料,以幫助未來的研究。 3. 盡量從 L-SP與 H-SP 兩者的生理意義來著手,多進行兩者的動力學實驗,包括外加 L78 能否使 H-SP 即具有 primer-independent 活性,且嘗試改變各反應物種的濃度來進行動力學量測。 |
|
| 07/06/14 |
7 何 杰 龍 (以 2-DE 篩檢 AIV 病毒蛋白質表現與修飾) |
|
1. AIV 抗體庫第三階段以合成 peptides 免疫的工作遇到三個主要的瓶頸: (1) 樣本過酸,免疫小鼠易夭折、(2)不易轉印,須改用 ELISA 或 dot blotting 方式篩選、(3) 合成之 peptides 品質不穩定。經討論後採取的對策如下:樣本先徹底抽乾以去除 TCA,再增加回溶的緩衝液濃度後進行免疫注射。以 N 端定序確定合成之 peptides 品質,後續的片段,要考慮找品質較穩定的廠商合成。 2. 要注意進行 paper search,了解目前關於 AIV HA 各種 post modifications 的研究現況。 3. 免疫 NP, M1, M2 peptides 的小鼠採血,對兩種亞型 AIV 效價測試的結果發現,H5 高於 H6,因其全部序列 homology 很高,須比對設計的片段是否剛好位於相異處。 4. 由 AIV deglycosylation 的結果顯示,2D 圖譜上 HA1 各蛋白質點均向下位移,但並無原先預測之狀況發生,因此判斷這些序列蛋白質點並非單純由 glycosylation 造成,須考慮 phosphorylation 的可能。待足夠 AIV 樣本到位,將研究焦點集中於此,並運用先前已篩出之 HA1 專一性單株抗體協助定位。 5. AIV deglycosylation 或其他 post modifications 的實驗須重複,最好有各階段的變化。 6. 思考這些蛋白質點位移與致病或生理機制之關聯,要與王老師實驗室密切討論。 7. 以目前篩出的單株抗體或採血,進行 AIV 2D 圖譜上各病毒結構蛋白質的初步追蹤,協助進行更精確的 LC/MS/MS 定序。 8. 加速進行 HA 與 NP 的 peptides 序列設計與合成。 |
|
| 07/05/31 |
17 王 信 傑 (Phytochelatin synthase 反應機制研究) |
|
1. 請確認目前有無 papers 發表利用 PC2/PC3 當基質的相關研究,或者反應機制 Plus-One- Rule 的提出。 2. 利用 HPLC 純化 PCn 的過程,10 分鐘 peak 應該加以定序確認身份,只靠位置比較推測為 GSSG 不可靠。 3. 除進行部分純化 PCS 2D 電泳外,另外也要跑 AtPCS 水解前後的 2D 電泳,以確認抗體辨識的片段。 4. 嘗試利用抗體製備 affinity column,將水解 AtPCS 片段或阿拉伯芥部分純化 PCS 通入 column 中,找出結合片段或內生性 PCS。Sample 可加以稀釋並反復注入管柱。 5. PC2/PC3 濃度要再提高,如此才能明顯看出對 PCS 活性的影響。 6. 在磷酸化實驗部分可針對基質添加順序加以探討,以便瞭解磷酸化過程。 7. 參考已發表之 PCS 模型與序列,推測可能進行磷酸化的位置,利用點突變方式來加以驗證。 8. 利用不同 truncated AtPCS 片段來進行 second binding site 與磷酸化研究,並導入酵素動力學實驗探討 PCS 作用機制。 9. 探討 PCS 磷酸化之生理意義,除設計不同 in vitro 實驗證明外,另導入對鎘敏感之細胞進行 in vivo 實驗。 |
|
| 07/05/24 |
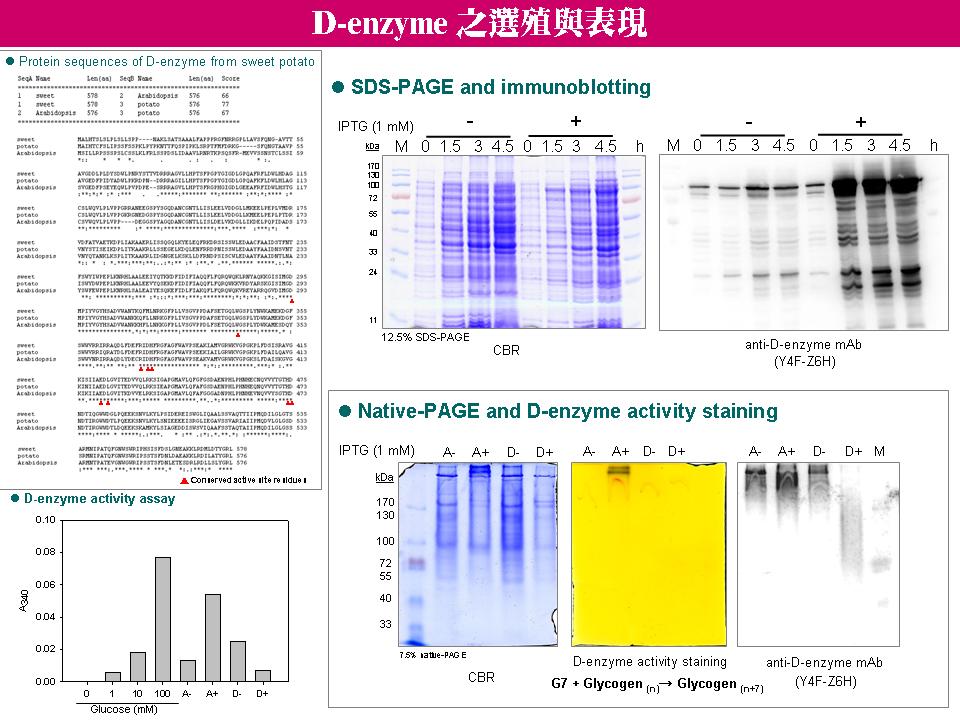
4 林 怡 岑 (L-SP, D-enzyme 與 proteasome 蛋白質間之結合與生理功能) |
|
1. 若懷疑細胞非單株,可再作一次 subcloning 來確認。 2. 需利用 SDS-PAGE 之 Western 來確認抗體的專一性。 3. 需確認是何種原因造成蛋白質在原態電泳中有 smear 的現象? 4. 重複利用 L-SP、D-enzyme 和 proteasome 之抗體,分別交互進行免疫共沉澱實驗,進一步確認 L-SP、proteasome 和 D-enzyme 之間的結合關係。 5. 確認蛋白質複合體的結合關係後,需進一步瞭解其所扮演的生理角色,最好能有免疫電顯或 confocal 之組織影像圖。 6. 以 L-SP 和 D-enzyme 之抗體分別進行 HX 之免疫共沉澱,所得到的蛋白質色帶強弱差太多,需設法解決此問題。 7. 若 protein A agarose 與抗體 cross-linking,不會影響抗體和抗原之間結合效果的話,以後可都改用 cross-linking 的方式進行免疫共沉澱實驗。 8. 免疫共沉澱所流洗出之蛋白質,應以 LC/MS/MS 進一步確定身份。 9. 比對 L-SP 和 D-enzyme 之高分子量色帶,應以 CBR 及活性染色圖譜為主。 10.比對 L-SP 和 D-enzyme 的序列相似度,看看兩者是否可能有相同的催化活性,或是具有類似的抗原決定基,而被相同的抗體所辨識。 11.目前認為 L-SP 與 D-enzyme 可能形成蛋白質複合體之證據:免疫共沉澱實驗、native / SDS 2D-PAGE 之次單元組成,以及高分子量活性色帶之相似性。 12. L-SP 可能受到不同磷酸化修飾而與不同蛋白質結合,因而扮演不同生理角色。 13.以醣蛋白染色法觀察蛋白質醣基化修飾作用,並確認 D-enzyme 是否為醣蛋白。 14.查閱 L-SP 和 D-enzyme 可能共同作用的相關文獻,使 L-SP 和 D-enzyme 構成蛋白質複合體之可能性更具有說服力。 |
|
| 07/05/17 |
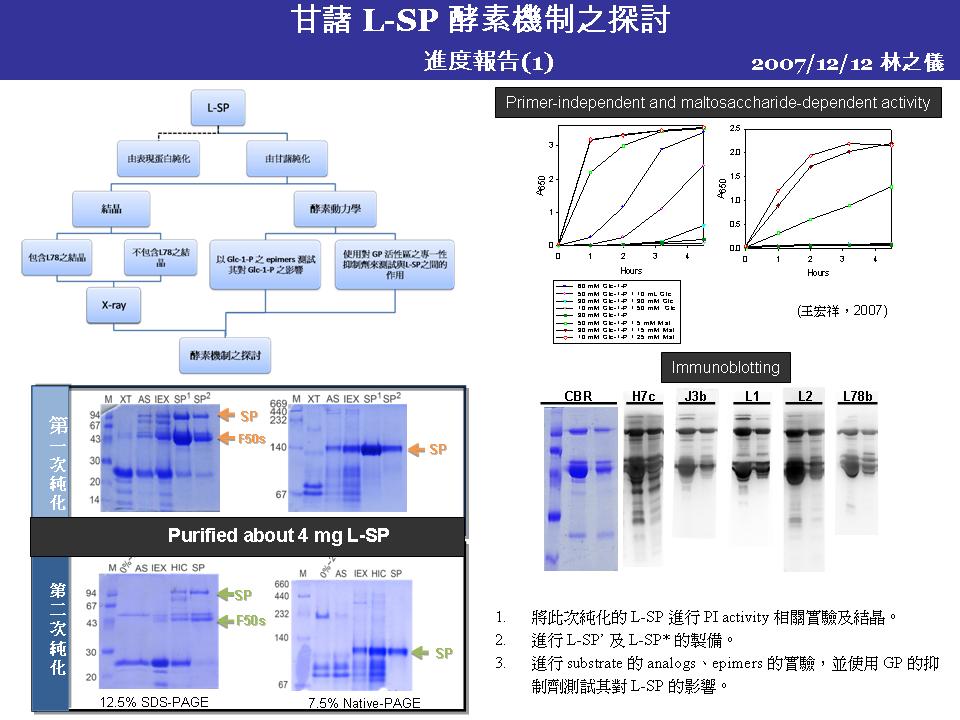
4 王 宏 祥 (L-SP primer-independent 活性之作用機制) |
|
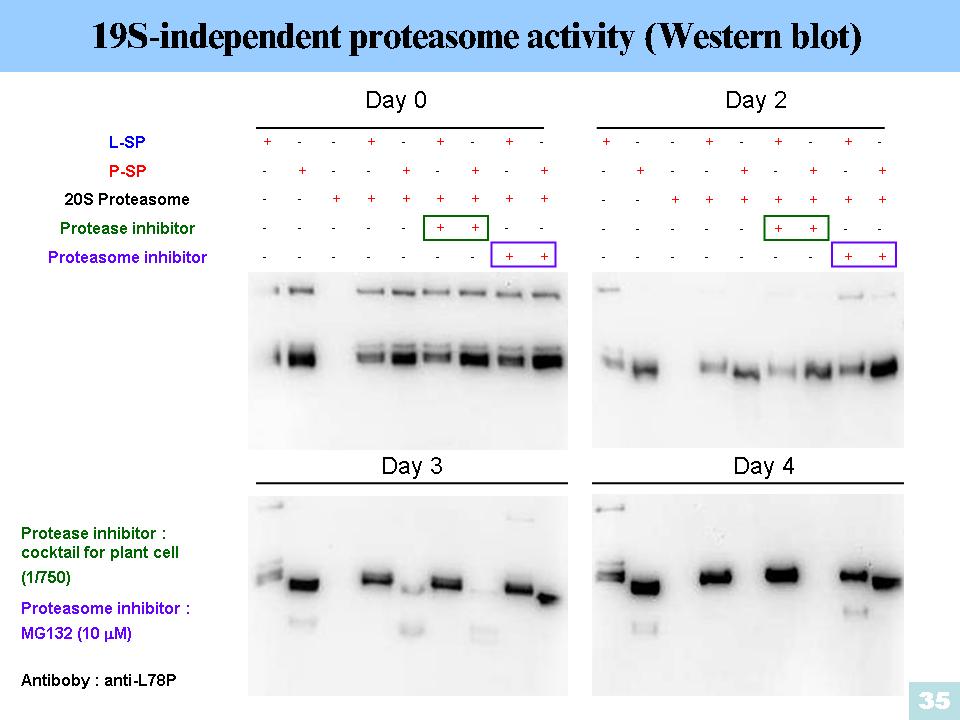
1. L78 與 ADPase 比對出來的 EKY 三個胺基酸,還須找出其 L-SP 結構上的相對位置,證實此三個胺基酸會與 Glc-1-P 作用。 2. L78 與 ADPase 比對出來的 EKY 三個胺基酸,附近序列的保守性不高,需要再多加 check。 3. L-SP PI activity 是為異位調控的實驗要再重複,同時外加 Glc 當 activator 的濃度太高,需往下調整。 4. L-SP 與 L-SP’ 從 G2~G7 的酵素動力學並沒有明顯差異。 5. 高濃度短鍊糖做為基質,會使碘液無法呈色,其中G3的現象較不尋常,需要從藥品品牌開始一步一步重新檢查。 6. 外加 proteasome 至磷酸化或沒有磷酸化的 L-SP,發現磷酸化的 L-SP 將解速度稍微比沒有磷酸化的來得快。 7. 根據上一面的實驗,分別再外加 proteasome 與 protease inhibitor 發現 proteasome inhibitor抑制效果比較好,但是此實驗必須改用 Western 再做一次。 |
|
| 07/04/12 |
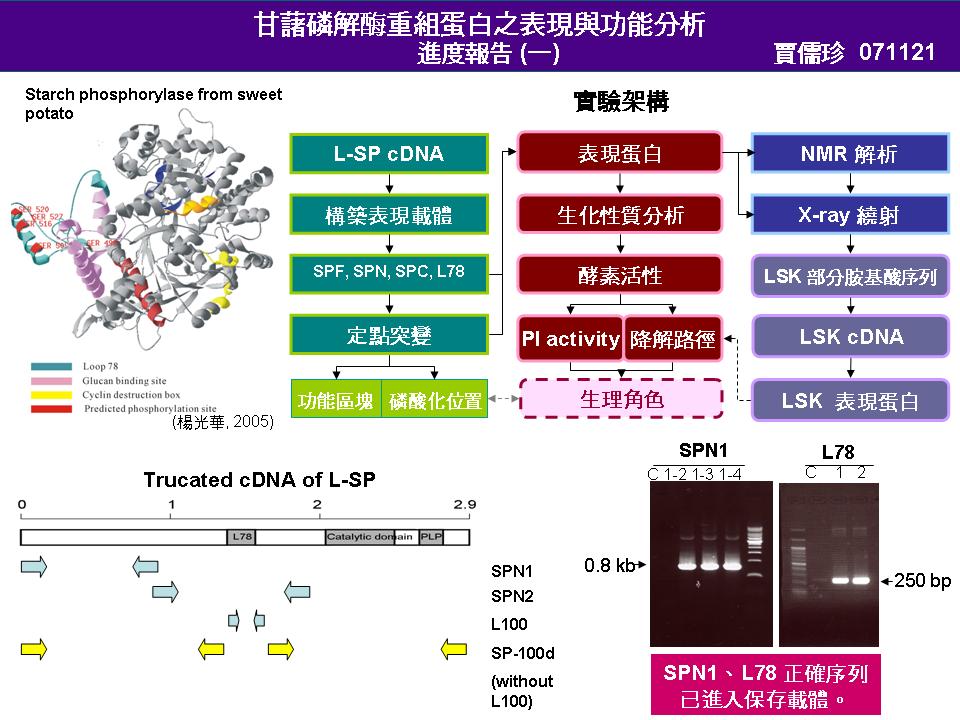
4 張 瓊 尹 (L-SP 蛋白質片段之表現與活性分析) |
|
1. 已成功表現出 C 端之 SPC,並做一些粗略的生化分析。 2. N 端 SPN 部分因為廠商做出的引子可能有缺陷,目前無法順利表現,但已經接入保存型載體,未來只要將其表現出來即可。 3. SP 全長部分已經接起來,但是因為保存載體與 SP 大小相近,無法將其分開,接入表現載體機率很低。對應之策略將用另一種限制酶將保存型載體切成兩段,則可與 SP 分開,增加接入表現載體機率。 4. 為何 J3B 抓不到純化出來的 F50? 抗體可能有問題,要確認。 5. 盡快將表現出的 SPC 做 N 端定序。 6. 為何活性染色沒有結果? 可能 SPC 之正電基團較多,導致蛋白質無法進入 Native gel,盡快預測其 pI 值。 7. 活性分析結果是否真的是 SPC 活性?是否有 phosphatase汙染?可加入 phosphatase 基質 PNPP (p-pitrophenyl phosphate) 測其活性即可知道。(但要考慮 SPC 也有 phosphatase 活性的情況) 8. SPC 可能只有磷解方向活性?因為這部份只有 A site,是否有做磷解方向之活性? 是有做過一次,但沒有成功,以後會再做。至於合成方向的活性只有測磷酸,而碘液呈色出不來,且也無法排除 phosphatase 污染,所以目前會盡快確定這些活性是否真的是 SPC 活性? 並確定是磷解或合成方向。 9. 有無外加 PLP ? 因為如果是合成方向,應該要有 PLP 存在 (磷解方向也要有 PLP)。或者可以加入 PLP inhibitor? 之後可以做 PLP binding site mutant。 沒有,因為當初認為細菌本身應該就會產生 PLP。 10. 轉入空 vector,表現後收一樣分劃位置的蛋白質,看是否還有水解磷酸之活性。 |
|
| 07/03/29 |
4 陳 均 全 (酵母菌 2476 鎘結合物質之身分鑑定) |
|
1. 要改善純化步驟: 經過膠體過濾、離子交換後的樣本,利用 FPLC 進一步純化,嘗試 Superose 6 和 Superdex 200 兩種膠體管柱,以 Superdex 200 效果較佳。經 FPLC 分離後,發現鎘高峰 (FCd) 和 UV 吸收高峰 (FPt) 分開。 2. 原態分子量測定: 因為 FPLC 分離再現性佳,因此利用 gel filtration standard 測定鎘結合物質的原態分子量,約為 9.6 kD,需再以較小範圍的標準品 (LMW gel filtration standard) 確定分子量。 3. 鎘結合物質身分鑑定 a. 醣蛋白質:利用 PAS 染色,分別以 IgG 及 BSA 為 positive 及 negative control,發現鎘結合物質 (FCd) 不呈色,推測不為醣蛋白質,也可能不隨電泳進入膠片中。以 glycosidase 測試是否為醣類物質,利用 a-glucosidase 及 a-mannosidase 反應,鎘結合物質之分子量及鎘結合量皆不受影響。 b. 核酸:分別利用 DNase 及 RNase A 處理,皆不受影響。 c. 蛋白質:以 trypsin、proteinase K、papain 等蛋白酶處理,皆不受影響。 因此,由以上各類測試,尚無法推斷鎘結合物質之本質為何。 4. 由上述 3c 推得鎘結合物質可能不是蛋白質,因此利用 PCI 容易將粗抽液中的蛋白質變性沉澱後,將水層部分進行膠體過濾管柱,發現其分子量及鎘結合量都不受影響,且除去大部分的蛋白質,經 HPLC 及 FPLC 確認 PCI 處理後的鎘結合物質 (PCICd) 和先前純化步驟的產物是相同的,且純度較佳。 5. 將 PCICd 以 TLC 分離後,再以 ninhydrin 呈色,發現呈現黃色而非胺基酸反應的藍紫色,因此推測並非 peptide 或胺基酸。應嘗試不同種類 TLC 材質或呈色法。 6. 未來主要以鎘結合物質身分之鑑定為主,利用元素組成分析、IR、NMR 等方法。 7. ATCC 2476 有將隔離子向外排出之機制,將觀察鎘結合物質的消長,討論在生理上所扮演角色。 |
|
| 07/01/22 |
Y 王 信 傑 (重金屬隔離機制之研究) 學位預備口試 (Y) |
|
1. 可嘗試用點突變方式來找出 PCS binding sites,作為直接證據。 2. 探討 PCS mechanism 要以重組蛋白為主,不要再從純化內生性 PCS 著手。 3. 請再確認金屬環是否真的與 PC 有關? ATCC2476 據了解並不產生 PC,但仍可產生金屬環,可見兩者關係並非絕對。 4. 論文題目中英文翻譯不符,請改正。 5. 盡快定出 H6 組成,推論出在細胞內的生理作用,這部分要趕快寫論文。 6. 抗體製備失敗部分應詳加檢討。PCS 催化模式與 papain 相似,但比對只秀出前端序列而已,應就全長序列加以比對,並注意有無可能 PCS 本身即是 protease。 7. 關於圖 1.12 鎘在酵母菌內的代謝途徑由於年代久遠,應該重新檢討一遍。例如鎘如何進入細胞內?進入後如何代謝等等? 8. 許多 legend 並沒有詳細說明實驗條件,應予已改善。 9. 在 PCS 重組蛋白純化部分,經 HisTrap column 純化完後電泳結果還有許多雜蛋白,應再經 FPLC 純化。 10.可嘗試用 GSH agarose gel 來純化經 proteinase K 作用後的 AtPCS 片段。 11.在進行抗體辨識抗原這部分實驗應重新檢討,可嘗試 sample buffer 中不加 2-mercaptoethanol。 12.可惜目前實驗都欠缺臨門一腳,未來的我應該學習找到事情重點,直接切入。 |
|
| 07/01/02 |
1 蔡 合 成 (綠竹筍抗體庫之應用) |
| 流 程 |
找出竹筍生長過程中,有消長蛋白質的相對抗體 ↓ 確定抗體所辨試抗原的身份 ↓ (1) 探討在竹筍生長過程中,此蛋白質扮演的功能 (2) 探討此蛋白質交互作用的蛋白質之功用及交互作用的功用
|
|
1. 先找出在竹筍生長過程中,在 2-DE 圖譜上有消長色點的抗原與抗體。 → 利用 2-DE Western 或晶片檢測方式,尋找目標抗體。 注意:單株抗體經冷凍乾燥後,可能會有問題,回溶時也必須相當小心。 2. 建議集合約十或二十株單株抗體混合之,以同時進行 2-DE Western 節省操作。 3. 晶片呈色機制構想:可將抗體點至晶片上,用竹筍全蛋白體與晶片上的抗體結合,再利用混合的抗體當作一抗,接著以二抗呈色。 4. IP 部分暫停,待上述計畫完成後,再來確定抗原身分。 5. 1DE Western 在 negative control 有二抗干擾,需要解決此問題。 未來工作: 1. 確定已製備好的單株抗體是否仍然可用,否則要重新生產抗體。 關 鍵 2. 找出在竹筍生長過程中,具消長蛋白質的身分及其相對抗體 (2-DE Western 或晶片)。 3. 嘗試利用晶片來篩選需要的抗體。 |
|
| 07/01/02 |
1 饒 驤 (L-SP 催化機制的假設與 H-SP相關研究) |
|
1. 從有機化學的觀點,分別探討在酸性催化和鹼性催化條件下短鏈糖合成的反應機制;並由已知反應物 (Glc-1-P) 和產物 (短鏈葡聚糖),及可能參與催化的機團 (pyridoxal phosphate 和兩個 lysine),及可能在合成過程中固定住短鏈糖的基團 (一個 lysine),提出一個假設的 L-SP 催化反應機制。 2. 先找出催化必需的殘基,方可進行下一步研究,所想到的方法有五種: (1) 利用表現出全長的 L-SP,將那三個 lysine 分別做 mutation,再看是否仍有催化活性,來判斷這三個 lysine 是否為催化所必需的殘基。 (2) 利用表現出 helix b 上的三個 lysine 被突變置換的 L-78,來進行 rescue,然後看是否仍有 rescue 的效果。 (3) 利用 X-ray 和 NMR 來直接研究 SP 和 單獨的 L-78 來獲得真實結構,可確定 helix b 是否真的為 helix。 (4) 利用合成 Glc-1-P 類似物,其能夠不可逆結合上 L-SP,然後進行結晶和 X-ray 繞射來直接觀察 Glc-1-P 周圍的結構,以判斷可能的催化所必須的 residues 為何。 (5) 利用研發出各種 G1P 類似物的合成方法,進行 SP 對各個抑制劑的動力學研究,進而研究 SP 活性區的結構和活性的關係。 但上述各方法目前皆有不宜之處,故暫且作罷。 3. 由於甘藷 H-SP 含量很少,故先以 H-SP 含量較多的植物進行初步分析。而選取四種植物 (甘藷、馬鈴薯、胡蘿蔔、菠菜) 進行 H-SP 含量比較,顯示馬鈴薯可能 H-SP 含量最多,但由於下列諸多因素使這次比較結果無法採信: (1) 植物樣本太少,所抽取的蛋白質太少,活性染色和活性分析結果不明顯。 (2) 由於各物種所含 H-SP 保守性不一,硫酸銨分劃應該要取 (0~100%),不該只取甘藷 H-SP 所在的分劃 (0~50%)。 (3) 操作過程太久,導致太多蛋白質降解。 關 鍵 (4) 其它技術不熟練的人為因素等。 未來工作: 1. 甘藷 L-SP 的含量很高,而甘藷 H-SP 的含量極少,卻對支鏈澱粉有極高的活性,嘗試深究其中的差異。 (應該已知是 L78 的關係) 2. 嘗試製備 H-SP 之單株抗體。 3. 嘗試利用 molecular cloning 的方法,表現 H-SP。 4. 嘗試定出 H-SP 的 DNA 及胺基酸序列,從分子構造的觀點,探究其與 L-SP 在生理上的角色及可能相互間的合作關係。 |
|
| 06/12/26 |
1 沈 志 昱 {母豬繁殖力相關之基因體與蛋白質體研究} |
|
2 ▼
|
1. 有無針對 development block 的研究? 搜尋相關的 paper。 2. 選定 peptide 時若有 Lys,可能在 map 時會產生額外的分支。已與合成 peptide 的公司確認過有技術使 Lys 不會產成影響。 3. 設計 peptide 時要注意轉譯後修飾如磷酸化、醣基化位置,要避免這些地方 (尤其是最強的)。 4. 除了從林恩仲老師提供之資料庫中選定一些目標基因外,也可從已發表的 paper 上找尋其他物種中,與早期胚胎發育相關之基因,特別針對 4-cell 到 8-cell 發育過程關鍵,再回來找豬的同源基因。如此雙管齊下,可增加找到關鍵目標基因的機會。 關 鍵 5. 不一定只看 4-cell 到 8-cell 階段,整個過程直到囊胚期都要檢查,但可花較多心力在 4-cell 到 8-cell。 6. 是否研究 4-cell 到 8-cell 就可以解決關鍵的胚胎發育問題? 7. 目前第一步先建立抗體庫,以 peptide 免疫製備抗體確定有效價後,須回歸 in vivo 胚胎上去確認身分,並繼續多方搜尋其他與繁殖力相關之基因。 8. 以 1D 電泳結果推測胚胎之蛋白質含量,發覺不足夠跑 2-DE (樣本量至少需目前的 20 倍)。未來確定身分時,若樣本數只夠作幾張 2D 該怎麼辦? 9. 將胚胎與鼠類 myeloma cell 融合的構想不可行,因 fusion 是靠機率,要有非常多數量的胚胎 (> 10 6)。 |
| 06/12/19 |
4 陳 均 全 {SP 2476 酵母菌的特殊抗鎘機制} |
|
|
1. SP2476 可在含有 1 mM CdCl2 環境下,生長不受到抑制,對鎘具有耐受性。 2. 將經鎘誘導 SP2476 粗抽液進行 RP C18 HPLC 分析,以 SP806 作為 positive control,發現 SP2476 粗抽液中不含有 PC2 或 PC3;以 AtPCS 和 SP806 粗抽液作為 positive control 進行 PCS 活性測試,SP2476 粗抽液中亦不含 PCS 活性。將 2476 經 Sephadex G-50 分離的鎘結合高峰,收集進行 RP C-18 HPLC 分析,亦不含 PC2 或 PC3 的訊號,由以上三實驗推測 SP2476 非利用 PC 結合鎘離子。 (實驗的起始點) 3. SP2476 鎘結合高峰幾乎不含有硫醇基,和 metallothionein 含有大量的硫醇基推論不符,且利用 metallothionein 的單株抗體在 SP2476 粗抽液電泳中,也偵測不到 metallothionein 存在。以上兩個實驗推測 SP2476 非利用 metallothionein 結合鎘離子。 4. SP806 及 SP2476 粗抽液的 Sephadex G-50 (C16×90 cm) 圖譜應以相同的條件 (相同膠體高度、蛋白質量和樣本體積) 重複實驗,確定 SP2476 鎘結合高峰,和 SP806 的 HMW, LMW PC 的原態分子量比較。 重 要 (要仔細比對) 5. 在 gel filtration (C26 × 90 cm) 的蛋白質樣本,添加 15~20% 甘油,可改善 Sephadex G-50 的分離效果,相對離子交換 (DEAE Sephacel) 的效果也較佳,故發現先前 LC/MS/MS 所做的鎘結合蛋白質身分鑑定可能有錯。 6. 蛋白質鎘結合能力之實驗必須重新設計流程,增加嚴謹度,避免非專一性吸附鎘所造成假象;先進行 DEAE 樣本經原態電泳分離,以固定距離割下,灰化測定鎘含量,確定鎘結合物質的泳動率位置。 (也要作好標準品比對) 7. 不要再侷限鎘結合物質為蛋白質的想法,以各種方法確認是否為 peptide、醣類,甚至核酸。 (一一以化學方法去檢測) 8. 在蛋白質粗抽液的二維圖譜上,SP2476 加鎘及不加鎘培養,沒有顯著誘導產生的蛋白質點;要再以軟體比對確認。 未來工作: 1. 首先確認 SP2476 鎘結合物質為何,再進行單株抗體製備。 重 要 (主要探索目標) 2. 由於 total protein 二維電泳圖譜,無法有效觀察蛋白質消長,擬分離液泡進行二維電泳分析。 (工程不小) |
| 06/12/12 |
4 王 宏 祥 (L-SP 的催化與降解機制) |
|
1. 投影片要標上頁碼。 2. 把 glycogen phosphorylase, H-SP 與 L-SP 序列在前面先比較, 並強調 L78 的獨特性。 3. L-SP, L-SP' 與 L-SP* 的示意圖要修改,因為 N-peptide 的 C terminal 胺基酸並不清楚斷到哪裡。 4. L-SP* 有少量 110 kD 的污染,所以 rescue 中的磷酸呈色會有活性。 5. L-SP 可以使用 Glc 當作 primer (L-SP' 則不行),是否 L78 上的酸性胺基酸可穩定 Glc 需再進一步的思考。(Glc 看起來反而有點像 allosteric inhibitor) 6. 雖然預測 Glc 沒有活性,仍必須補做一個只加 Glc 不加 Glc-1-P 的 control 組。 7. 糖電泳及染色方法,是先將糖分子上的半縮醛標定螢光,再進行電泳,但是 Glc-1-P 上已無半縮醛,應該看不到其訊號。 8. 短鍊糖的實驗是否為 b-amylase 汙染所觀察到的假象,可利用外加氯化汞的方式再做一次。 9. 雖然預測 Glc 沒有活性,但仍要補做 Glc 的的酵素動力學。 10. 雙倒數作圖中,以磷酸呈色值作圖會比碘液呈色線性好些。 11. 對 L-SP' 而言,最短可運用的 primer 為雙糖。 結 論? (G3 的結果有點奇怪) 12. L-SP 與 L-SP' 各種短鍊糖的動力學 Km 交叉點,似乎可交在同一點,證明從雙糖開始無論是否有 L78 都不影響其親和力,要進一步說明 G1 與 G2 以上的 oligosaccharides 為不同的 binding site。 (可以切入) 13. 磷酸化對斷裂的影響必須再重複多做幾次。 未來工作: 1. 重新製備 L-SP*,並建立標準製備流程。 (加緊進行) 2. 重複 rescue 的實驗,並注意外加入 L78 的濃度比例,另外要補做各濃度的 L78 先加熱後再外加的對照組。 3. 探索 L-SP 的斷裂機制。 重 要 (應該列為主要探索目標) |
|
| 06/12/05 |
3 林 怡 岑 (L-SP, D-enzyme 與 proteasome 的關係) |
|
1. 瞭解 Tetlow 是如何設計實驗,證明 SP 和 SBE 形成蛋白質複合體後,所扮演的生理角色。 2. 利用 D-enzyme 和 proteasome 之抗體分別進行免疫共沉澱實驗,進一步確認 L-SP, proteasome 和 D-enzyme 之間的結合關係。 重 要 3. 需進一步探討 L-SP 是否有其他磷酸化位置? 可用表現的 L-SP 去進行磷酸化實驗,防止內生性磷酸化的干擾。 重 要 (可以切入) 4. 若 proteasome 和 L-SP 結合,是否會影響 L-SP 的活性? 5. 以 HX 蛋白質混合物進行酵素動力學的結果意義不大。 6. 比對 L-SP 和 D-enzyme 的序列相似度。 7. 注意 L-SP, proteasome 和 D-enzyme 形成的蛋白質複合物,是否只是 in vitro 聚集的假象 (reconstitute 混合實驗)。 8. 參考 Plant J. 由 S. Ball 發表的有關綠藻 starch phosphorylase 和澱粉合成有關的 paper。 9. L-SP 和 D-enzyme 是否可能有相同的催化活性? 10. 可改用 Sephacryl S-500 進行 HX 等高分子量蛋白質之純化。 11. 可利用 Hydroxylapatite 及 D-enzyme 抗體進一步純化及分析 D-enzyme。 12. 純化 D-enzyme 的過程中,都無法有效地與 L-SP 分離,可見 in vitro 混合 L-SP 和 D-enzyme可能無法產生 HX,而需要其他的修飾作用。 (怎麼說?) 13. 需注意有效數字之表示方式,並善加利用符號作有效的表達。 14. 可利用 SDS-PAGE 觀察蛋白質磷酸化與否之改變。 15. 實驗設計上,實驗組與控制組的變因只能有一個。例如,控制組應該也要添加鎂離子,或加熱失活的 CIAP,另外,外加 ATP 需注意 pH 的變化。 16. 為何 L-SP 對 starch 的親和力較高,而 D-enzyme 對 glycogen 的親和力較高? |
|
| 06/11/21 |
6 何 杰 龍 (以 peptide bank 製備 AIV 病毒的 mAb bank) |
|
1. 對於實驗要更積極一點,考慮太多也許不是件好事,趕快找到目標後就要衝。 2. 必須鞏固既有的 HA 單株抗體部分,詳細去確定已經得到的各單株特性。 3. 使用太多複雜的方法預測 peptides 的免疫性,但結果很多互相矛盾,最後還是不知道要選擇哪一段。希望先選用 2~3 種軟體,分析 AIV 中全部 10 種蛋白質中的 3~4 種,這些工作要盡快完成。 重 要 4. 如何確定這些預測方法的可信度? 會不會預測出來都是錯誤的結果? 一般而言,這些預測方法,雖然無法達到 100%,但應該有一定的可信度。而且我選擇片段的標準,是以功能與重要性為主,預測的免疫性為輔,如果是非常重要的片段,再怎樣都要想辦法進行免疫。 5. 以現有的 HA 進行 LC/MS/MS 片段進行 antigenicity 預測方法的驗證,在邏輯思考上有些問題,這並不能證明這個片段就是該單株抗體辨識的 epitopes。若是這個基礎不能成立,以該方法預測的結果較不可靠。 6. 因 H5N1 AIV 的相關研究較多,將已知 H5N1 的片段進行預測,並與 3233 或 2838K 的片段比對,應該更可以印證其準確度。 重 要 (可以切入) 7. Epitopes bank 應改為 epitopic antibody bank 較為適當。 8. 根據本實驗室以 peptides 製備單株抗體的經驗,依片段抗原性不同及小鼠的個別差異,成功率約為 1/3,因此每一個蛋白質選擇至少 3 個免疫性較高的片段,依循高產能抗體庫製備流程『一起免疫分別篩選』的原則建立抗體庫,理論上可篩到針對所有蛋白質的單株抗體。 9. 要考慮 glycosylation 與 phosphorylation 的位置。 |
|

{kind=link}